Kalıtsal olgularda otosomal dominant geçiş niteliği vardır. Olguların bir bölümünde kromozom anomalisi saptanır. Prepubertal gelişme geriliği bebeklerde ve çocuklarda göze çarpan ilk bulgulardan biridir. Brakisefali, yüksek ve geniş alın bombesi, laterale kaymış kaş çıkıntıları dikkat çekicidir. Epikantus, derin orbitalar, strabismus, myopi, retina dekolmanı gibi göz bulgularının yanı sıra işitme sorunlarına yol açan orta kulak infeksiyonları ile kulak kepçesi malformasyonları izlenir. Geniş ve yuvarlak yüzde orta bölüm hipoplaziktir, yanaklar dolgundur. Filtrum kısadır. Kalın dudaklar ve üst dudak yarığı görülür. Çeneler ve ağız bulguları arasında mikrognati, damak yarığı, hipodonti, taurodontism, bruksizm önemlidir. Beyin anomalileri, nöropati bulguları, davranış bozuklukları, uyku bozuklukları, hipotoni, zeka geriliği gibi nöropsikiyatrik bulguları ile konjenital kalp defektleri ve skolyoz öteki bulgulardır.
Dubowitz sendromu, ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur; genel bir gelişme geriliği vardır.
Oral-Facial-Digital sendrom II (orofaciodigital sendrom 2, Mohr sendromu; Mohr-Claussen sendromu), ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur. Belirgin bir genel gelişme geriliği saptanır.
Ağız-Yüz-Parmak sendromu tip 6 , ektodermal displazi bulguları da içeren, otosomal resesif geçen, ender görülen kalıtsal bir sendromdur. Belirgin bir genel gelişme geriliği saptanır.
OPD sendromu (otopalatodigital sendrom), X-kromozomu aracılığıyla dominant (XLD) yolla geçen kalıtsal bir sendromdur (Gorlin-Cohen sendromu hariç). Kız çocukları daha hafif etkilenir. Tümü benzer bulgular içeren 5 tipi vardır:
- Otopalatodigital sendrom tip I (OPD tip1)
- Otopalatodigital sendrom tip II (OPD tip2)
- Gorlin-Cohen sendromu (ilgili sayfaya gidiniz)
- Melnick-Needles sendromu (tip 1 ile allelik bağlantısı vardır; ilgili sayfaya gidiniz)
- Terminal osseöz displazi ve pigmentli deri defektleri (çok enderdir, ayrıntı verilmemiştir).
Gorlin-Cohen sendromu , ektodermal displazi bulgularını da içerebilen bir OPD sendromu tipidir. 2 fenotipi vardır:
- Frontometaphyseal dysplasia 1
- Frontometaphyseal dysplasia 2

Baraitser-Winter sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; 2 fenotipi vardır. Genel gelişme geriliği izlenir. Kraniyosinostoz nedenli trigonosefali ve mikrosefali saptanır. Boyun kısadır. Hipertelorizm saptanır, kaş çıkıntıları yüksektedir. Göz kapakları iridir ve bilateral ptozis saptanır. Mikroftalmi ile iris ve retina defektleri (koloboma) izlenir. Kulak kepçeleri küçüktür ve aşağıdadır; işitme sorunları vardır. Burun sırtı kalın, ucu büyüktür. Uzun bir filtrum altında ince bir üst dudak bulunur. Alt dudak kalındır. Üst dudak ve damak yarıktır. Altçene geridedir (retrognati). Omurga sisteminde kifoz ve skolyoz türü malformasyonlar saptanır. Başparmak sayısı birden fazladır (polidaktili). Değişik oranlarda konjenital kalp anomalileri ve ürogenital anomaliler görülür. Beyinde corpus callosum anomalileri ile frontal lob anomalisi saptanır; epilepsi ve zeka geriliği bulguları vardır. Genel hipotoninin yanı sıra omuz kaslarında giderek artan güçsüzlük görülür. Deri lenfoması ve lösemi riski yüksektir.
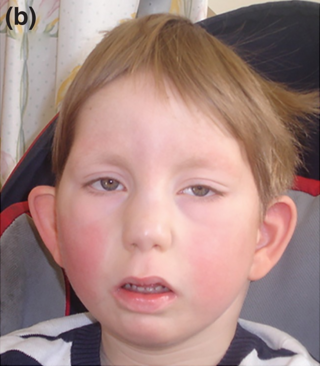
Au-Kline sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; böbrek, kalp ve yüz bulgularıyla öne çıkar.

Emanuel sendromu, t(11;22)(q23;q11.2) ayrımı bozukluğu olarak tanımlanan bir kromozom anomalisi sendromudur.
Frontonazal displazi, otosomal resesif yolla aktarılan, alın ve burun bölgelerinin malformasyonlarıyla karakterize kalıtsal bir sendromdur; ALX3 genindeki mutasyonun sonucudur. En önemli bulgu, frontal kemikte ve yüz kemiklerinde orta çizgi yarıklarının bulunmasıdır ile bunun sonucunda ortaya çıkan nazofrontal ensefalosel’dir. Alın derisinde lipoma olabilir. Mikroftalmi saptanır, gözler birbirinden aşırı uzaktadır (hipertelorizm); ptozis, kapak bileşkelerinde anomaliler, katarakt ve epibulbar dermoidler belirlenir. Kulak kepçeleri aşağıdadır, işitme sorunları vardır. Burun kökü aşırı yayvandır; bu alanda saptanan malformasyonların en önemlisi yarıktır. Üstçenede, paranazal sinüslerde ve frontal sinüslerde hipoplazi izlenir. Kalpte Fallot tetralojisi saptanabilir. Pektoral kaslar hipoplazi ya da agenez olabilir. El parmakları kısa ve kıvrıktır. Beyinde Corpus callosum anomalileri saptanır, zeka geriliği vardır. Akromelik frontonazal disostoz fenotipindeki bulgular daha azdır.

IFAP sendromu, X-kromozomu aracılığıyla resesif (XLR) olarak aktarılan kalıtsal bir sendromdur, özellikle erkek çocukları etkilenir. Oligohidramnios başlangıç noktasıdır. Gelişme geriliği ve mikrosefali belirgindir. Alın bombelidir. Deride ektodermal displazi bulguları olan alopesi, kaş ve kirpik yokluğu ile küçük tümsekler oluşturan iktiyoz (pullanma) ve ekzama türü lezyonlar vardır. Tırnak yapısı bozuktur. Gözlerde, kornea ülserleri ve opasiteleri ile keratit nedenli fotofobi görülür. Kulaklar iridir, işitme sorunları olabilir. Yarık damak saptanır.

Kabuki sendromu, iki fenotipi olan bir sendromdur: Kabuki sendromu 1 ve Kabuki sendromu 2 (Kabuk2). Kabuk1, otosomal dominant yolla aktarılan kalıtsal ya da izole olgular biçiminde görülür; Kabuk2 ise, X-kromozomu aracılığıyla dominant yolla (XLD) aktarılır. Her iki fenotipini çok sayıda ortak bulguları vardır; özellikle yüz, iskelet sistemi, gelişme geriliği, parmakizleriyle ilgili bulgular ve zeka geriliği önemlidir.
Koolen-de Vries sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Güçlü zeka geriliği sendromlarının önemli bir fenotipidir. Cüceliğe yakın düzeyde gelişme geriliği vardır. Yüz uzun, alın ve yanaklar geniştir. Kulaklar kıvrımlıdır. Gözlerde irisler soluktur; şaşılık, ptozis ve kapak yapışıklıkları izlenir. Yüksek ve silindirik burun sırtının altında armut biçiminde bir burun ucu vardır. Alt dudak sarkıktır. Dar ve çukur damakta çoğu hastada yarık da saptanır. Dişler küçüktür (mikrodonti). Kardiyovasüler sistemde, kalpte septum defektleri, pulmoner kapak stenozu ve aorta anomalileri belirlenir. Meme uçları birbirinden uzaktır. Hidronefroz, üriner sistemde sık görülen bir patolojidir.

Mowat-Wilson sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Goldberg-Shprintzen sendromu ile çok sayıda ortak bulgusu vardır. Bunlar arasında mikrosefali, psikomotor gerilik, hipotoni, zeka geriliği ve epilepsi en önemlileridir.

MVA sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. 3 fenotipi vardır; fenotip 1 en sık görülenidir. Anöploidi olarak niterlendirilen kromozom sayısı anomalisinden kökenlidir; kromozom eksikliği (monosomi) ya da fazlalığı (trisomi) vardır. Ancak, bazı hücrelerdeki kromozom sayısının normal olması nedeniyle “mozaisizm” grubu içinde yer alır.

Pena-Shokeir sendromu II, otosomal resesif yolla aktarılan, serebrookülofasiyoskeletal sendrom grubundan, kalıtsal bir sendromdur. Cockayne sendromu tip II'nin fenotipi olabilir. Mikrosefali, zeka geriliği, garip yüz yapısı, doğumsal katarakt ve artrogripozis bulgularının ön planda olduğu bir tablodur. İlk 2 yaş içinde yüksek ölüm riski vardır.

Toriello-Carey sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Doğum ölçümleri normaldir, ancak zamanla gelişme geriliği bulguları belirir.

Catel-Manzke sendromu, Pierre Robin sequence bulgularına ek olarak işaret parmaklarında, ikinci metakarp ile proksimal falanks arasına eklenmiş artı bir kemik nedeniyle ortaya çıkan bir malformasyon içeren, çoğu spontan gen mutasyonuyla izole olgular biçiminde ortaya çıkan bir sendromdur. Otosomal resesif yolla aktarılan birkaç kalıtsal olgu da bildirilmiştir.
CHARGE sendromu ;, CHD7 genindeki spontan mutasyona bağlı izole olgulardır; az sayıda otosomal dominant yolla aktarılan kalıtsal örnekler de vardır. Gözlerdeki yapısal bozukluklar, kalp defektleri, üst solunum yollarındaki darlıklar, gelişme geriliği, genital hipoplaziler, kulak ve ekstremite anomalileri ön plandaki bulgulardır.
Opitz-Kaveggia sendromu, X-kromozomu aracılığıyla resesif yolla (XLR) aktarılan kalıtsal bir sendromdur. Zeka geriliğinin yanı sıra, kafatası ve parmaklarla ilgili bulgular ön plandadır. Fiziksel gelişme geriliği nedeniyle boy kısadır. Makrosefali ve asimetrik kafatası (plagiosefali) vardır; ön bıngıldak geniştir ve geç kapanır. Saçlar ince ve seyrektir. Öndeki saçlar yukarı doğru toplanmıştır. Yüz derisi kırışıktır. Alın bombesi yüksektir. Boyun kısadır. Kulaklar küçüktür ve işitme sorunları vardır. Gözler çekiktir, birbirlerine olan uzaklıkları artmıştır (hipertelorizm); strabismus izlenir. Burun belirgin, filtrum uzundur. Ağız büyüktür (makrostomi), ancak altçene küçüktür (mikrognati). Alt dudak iridir; üst dudak ve damak yarığı vardır. Damak dar olduğu için çiğneme düzlemi bozuktur (maloklüzyon). Burun boşluklarında darlıklar vardır.
