
DiGeorge sendromu, 22q11 deletion içeren sendromlar grubunun bir fenotipidir. Olguların bir bölümü kalıtsaldır, otosomal dominant yolla aktarılır.Teratojenlerin ve diabetes mellitus'un neden olduğu mutasyonların da etkisi önemsenmektedir. Gebeliğin 4-7. haftasında beliren 3.-4. faringeal ark kompleksi etkilenmesine bağlı konjenital anomalilerle karakterizedir.

Coffin-Siris sendromu, ektodermal displazi bulgularını da içeren, otosomal dominant geçen kalıtsal bir sendromdur. 8 fenotipi vardır. Çene-yüz bulguları fenotiplerden 3'ünde belirgindir. Hastalarda genel gelişme geriliği vardır. Kafatası küçüktür (mikrosefali).
Kraniyoektodermal displazi (Cranioectodermal dysplasia), ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur. 4 fenotipi vardır:
- Cranioectodermal dysplasia 1 (Sensenbrenner sendromu; Levin sendromu; arthrodentoosseous dysplasia)
- Cranioectodermal dysplasia 2
- Cranioectodermal dysplasia 3
- Cranioectodermal dysplasia 4
Gorlin-Cohen sendromu , ektodermal displazi bulgularını da içerebilen bir OPD sendromu tipidir. 2 fenotipi vardır:
- Frontometaphyseal dysplasia 1
- Frontometaphyseal dysplasia 2
Even-Plus sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Genel gelişme geriliği vardır. Yenidoğanda, deri malformasyonu saptanır. Brakisefalik kafa biçimindeki kafatasında saçlar seyrektir. Boyun kısa, kaş çıkıntıları belirgindir. Kaşlar birbirine birleşmiş olabilir. Kulak deliği yoktur ya da dardır (microtia). Burun hipoplaziktir, ucunda yarık vardır. Yüz orta bölüm hipoplazisi nedeniyle üstçene küçüktür (mikrognati), damak çukuru belirgindir. Üst çenede tek orta kesici (santral) diş vardır, lateral dişler eksiktir (hipodonti). Kalpte konjenital anomaliler saptanır. Anüste darlık olabilir. Üriner sistemde, böbrek hipoplazisi ve infeksiyonlar olabilir. İskelet sisteminde, omurga yarıkları ve koksiks agenezi saptanır; uzun kemiklerde malformasyonlar vardır. Atopik dermatit olabilir. Corpus callosum agenezi, beyinde saptanan en önemli anomalidir.
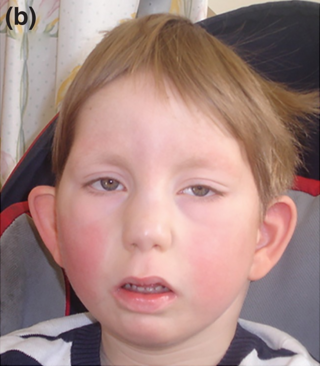
Au-Kline sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; böbrek, kalp ve yüz bulgularıyla öne çıkar.

Emanuel sendromu, t(11;22)(q23;q11.2) ayrımı bozukluğu olarak tanımlanan bir kromozom anomalisi sendromudur.
Galloway-Mowat sendromu, kalıtsal bir sendromdur. 8 fenotipinin 7'si otosomal resesif yolla aktarılır. Genel gelişme geriliği nedeniyle boy kısadır. Mikrosefali ve küçük bir yüz yapısı saptanır; kafa yanlardan basıktır, alın bombesi yüksektir. Fenotiplerin bir bölümün saptanan çok sayıdaki göz anomalisi nedeniyle görme kusurları belirlenir. Dış kulak ve burun malformasyonları olabilir. Altçene küçüktür (mikrognati), ağız açıklığı malformasyonları ve çukur damak görülebilir. Ağız bulgularının bir bölümü fenotipe özgüdür: mikrodonti, yarık damak, mine hipoplazisi saptanır. Fenotip 4 ve 5'te oral bulgu yoktur.

Joubert sendromu, kalıtsal bir sendromdur; 36 fenotipinden 34'ü otosomal resesif yolla aktarılır, olguların bir bölümü Meckel sendromu ile çakışmaktadır. Joubert sendromunun tüm fenotiplerinde 3 temel bulgu vardır: santral sinir sistemi anomalileri, fizik gelişme geriliği, solunum sorunları. Serebellum (beyincik) vermisi ve beyin sapı hipoplazisi ile beyin radyolojisinde saptanan “azıdişi bulgusu ” santral sinir sistemi anomalilerinin başında gelir. Ataksi ve hipotoni sık görülür. Bazı hastalarda oksipital meningosel ya da meningomyelosel olabilir. Schinzel acrocallosal sendromu, önemli fenotiplerinden biridir.
Mikroftalmi sendromları, 18 fenotipi olan, etkilenen gen türüne göre farklı yollarla -otosomal dominant (AD), otosomal resesif (AR), X-kromozomu dominant (XLD), X-kromozomu resesif (XLR)- aktarılan kalıtsal patolojilerdir. Ortak bulgular yanı sıra farklı sistemlere özgü bulgularla da karşılaşılmaktadır. Ortak bulguların en büyük kümesi gözlerle ilgilidir.
C sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Bohring-Opitz sendromu, C sendromu'nun fenotipi olarak benimsenir.
Multipl konjenital anomaliler-hipotoni-epilepsi sendromu, 3 fenotipi olan kalıtsal bir sendromdur. Fenotip 1 (MCAHS1) ve fenotip 3 (MCAHS3) otosomal resesif, tip 2 (MCAHS2) ise x-kromozomu aracılığı ile resesif olarak aktarılır. Birçok enzimin çalışabilmesinde, kompleman sisteminin düzenlenmesinde ve hücrelerarası iletişimin sağlanmasında önemli katkıları olan, fosfogliserid yapısındaki glikosilfosfatidilinositol sentezindeki sorunlarından kökenlidir. Nörolojik belirtiler sendromun fenotiplerindeki temel bulguları oluştururlar; bunlara eklenen çok sayıdaki anomalilerin yeri, sayısı ve gücü farklıdır.

Schinzel-Giedion sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Güçlü bir zeka geriliği ile birlikte garip bir yüz yapısı ve çok sayıda doğumsal anomaliler görülür. Cüceliğe yakın düzeyde boy kısalığına neden olan genel gelişme geriliği vardır.
Ritscher-Schinzel sendromu, 2 fenotipi olan kalıtsal bir sendromdur.
Seckel sendromu, 9 fenotipi olan, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Doğumdan önce belirlenen ve yaşam boyu süren, cücelik düzeyinde gelişme geriliği ile kuş kafasını andıran yüz görünümü, mikrosefali ve zeka geriliği saptanır. Alın dardır ve geriye çekilmiş gibidir. Gözler fırlaktır; iki taraflı optik atrofi nedeniyle görme sorunları yaşarlar; gözler çekiktir, kapaklarında yapışıklıklar vardır. Stabismus olabilir. Burun kuş gagasını çağrıştırır. Kulaklar biçimsizdir, kulak memesi yoktur ve işitme güçlüğü belirlenir. Asimetrik bir yüzde, altçene küçüktür ve geridedir (mikroretrognati), damak çukurdur ve yarık saptanır. Dişler küçüktür (mikrodonti) ve eksiktir (hipodonti); bu nedenlerle, çiğneme düzlemi bozuktur (maloklüzyon). Mine hipoplazisi belirgindir.
Toriello-Carey sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Doğum ölçümleri normaldir, ancak zamanla gelişme geriliği bulguları belirir.

Bohring-Opitz sendromu, otosomal dominant yolla aktarılan, gelişme ve zeka geriliği bulgularının ön planda olduğu, hastaların çoğunun çocukluk yaşlarında kaybedildiği bir sendromdur. C sendromu'nun fenotipi olarak benimsenir.

MEHMO sendromu, X-kromozomu aracılığıyla resesif (XLR) yolla aktarılan bir sendromdur; 50'nin üzerindeki “zeka geriliği” sendromlar kümesinin bir elemanıdır.

Saethre-Chotzen sendromu (acrocephalosyndactylia III), fiziksel gelişme geriliği, kraniyosinostoz nedenli kafatası anomalileri, asimetrik yüz, göz ve parmak malformasyonlarının saptandığı otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Kraniyosinostozun çok sayıda eklemi etkilediği olgularda “kafaiçi basıncı artışı sendromu (KİBAS)” gelişebilir.

Simpson-Golabi-Behmel sendromu, X-kromozomu aracılığıyla resesif olarak (XLR) aktarılan kalıtsal bir sendromdur; 2 fenotipi vardır.
