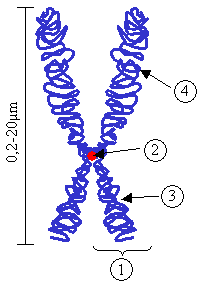
Kromozom, ; DNA'nın "histon" proteinleri etrafına sarılmasıyla, yoğunlaşarak oluşturduğu, canlılarda kalıtımı sağlayan genetik birimlerdir. Kromozomlar mikrometre boyutunda olup hücre bölünmesinin metafaz aşamasında ışık mikroskobu ile görüntülenebilmektedirler.

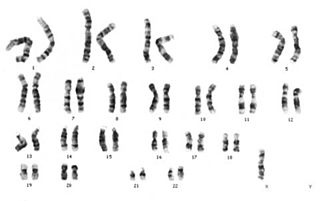
Karyotip, bir hücredeki kromozomların özdeş çift kromozomlar halinde eşlendikten sonra belli bir düzene göre sıralanmasıdır.
Cinsiyet, erillik ve dişilik arasında farklılık gösteren özellikler aralığı veya bağlama göre, bu özellikler biyolojik cinsiyeti ve cinsiyete dayalı toplumsal yapıları kapsayabilir.
Kromozom karyotiplemesinde günümüzde beş özellik incelenmekteir.
- Uzunluk, ayrı ayrı veya toplam kol uzunlukları,
- Sentromer konumu, buna göre telomerler ve kromozom şekli belirlenir.
- Sekonder boğumun bulunup bulunmaması ve varsa konumu,
- Kromozomların bant özellikleri, Giemsa ve fleurosans boyalarla aynı gruptaki kromozomların tayininde kullanılır.
- Otoradyografik özellikler, radyoizotop maddelerle işaretlenmiş ve nukleotidlerin kalıtımı ve DNA içi tekrarları belirlenebilir.

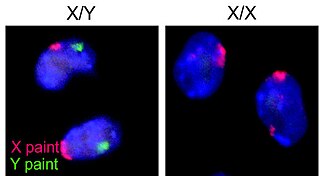
X kromozomu, iki eşey kromozomundan biridir.
Y kromozomu, iki eşey kromozomundan biridir. Bütün erkeklerde normalde "44, XY" normal karyotip olarak X kromozomuya birlikte bulunur.

Down sendromu, trizomi 21 ya da mongolizm; genetik düzensizlik sonucu insanın 21. kromozom çiftinde fazladan bir kromozom bulunması durumu ve bunun sonucu olarak ortaya çıkan genetik bir bozukluktur. Down sendromu, bireyin 1 yaşından daha uzun süre yaşayabildiği tek otozomal trizomidir.
Turner sendromu, kromozom anomalisi sonucu ortaya çıkan ve kız çocuklarını etkileyen bir sendromdur; bir dişide eşey kromozomlarından birinin bulunmaması sonucu ortaya çıkar. Turner sendromluların fenotipi dişi olarak görülür fakat; eşey organları ve eşey hücreleri gelişmez. Kısır bireylerdir.

Klinefelter sendromu ya da 47, XXY sendromu; hücre bölünmesi sırasında, eşeysel kromozom düzensizliklerinden kaynaklanan semptomların kişide görülmesi durumudur.
XYY sendromu veya süper erkek sendromu; erkek bireyin iki Y kromozomu taşımasına neden olan bir eşey kromozomlarında meydana gelen anöplodi durumudur.
X inaktivasyonu, dişi memeli hücrelerinde iki adet bulunan X kromozomlarından birinin inaktive edilmesi işlemi. İnaktive edilecek X kromozomu, baskılayıcı heterokromatin ile paketlenerek, bu kromozomun üzerinde bulunan genlerin ifade edilmesi önlenir. Böylece, sadece bir X kromozomuna sahip erkeklerle, iki X kromozomuna sahip dişiler arasındaki X kromozomu dengesi sağlanmış olur. İnsan ve fare gibi gelişmiş memelilerde, inaktive edilecek kromozomun seçimi rastgele yapılır ve inaktive edilmiş X kromozomu, içinde bulunduğu hücrenin yaşamı boyunca inaktif olarak kalır
Trizomi, diploit sağlıklı canlılarda normalde iki olması gereken kromozom sayısının üç tane olmasıyla bilinen anöploidi şekillerinden biridir. En çok bilinen şekli 21. kromozomun trizomisi olan Down sendromudur.

İnsanlar erkek ve dişi olmak üzere iki cinsiyette bulunurlar. Her insan 46 kromozomdan oluşur. Bunlar erkeklerde 44+XY dişilerde ise 44+XX olarak bulunur. Çiftleşme sırasında anne sabit X kromozomunu verir baba ise %50 oranla ya X ya da Y kromozomunu verir. Böylece cinsiyet belli olur.
Genetik bozukluk, genlerde ve kromozomlarda görülen anomaliler sonucu ortaya çıkan durumdur. Kanser gibi bazı hastalıklar yaşam sırasında edinilen ve bazı hücrelerde görülen genetik anomaliler nedeniyle olsa da "genetik hastalık" terimi genellikle vücuttaki tüm hücrelerde bulunan ve döllenmeden beri var olan hastalıklar için kullanılır. Bazı genetik bozukluklar, sperm ve yumurtalar gibi üreme hücrelerini oluşturan mayoz bölünme sırasında oluşan kromozom anomalileri nedeniyle ortaya çıkar. Bunlara örnek olarak Down sendromu, Turner sendromu (45X0) ve Klinefelter sendromu sayılabilir. Diğer genetik değişiklikler ebeveynler tarafından tohum hücrelerin oluşturulması sırasında ortaya çıkabilir. Bunlara bir örnek frajil X sendromu ya da Huntington hastalığına neden olabilen üçlü yayılma tekrar mutasyonudur. Hatalı genler ebeveynlerden olduğu gibi alınmış da olabilir. Bu genellikle sağlıklı ama resesif gen taşıyan iki kişinin üremesi ya da hatalı genin dominant olması sonucunda olabilir.
Rett sendromu, yaygın gelişimsel bozukluklardan birisi olarak sınıflandırılan beyinsel gelişim bozukluğudur. Ancak bunun yanlış bir sınıflandırma olduğunu ve benzer şekilde otistik belirtiler gösteren frajil X sendromu, tüberoz skleroz ya da Down sendromunun yaygın gelişimsel bozukluklar olarak sınıflandırılabileceğini önesüren görüşler bulunmaktadır. Bu sendromun belirtileri kolaylıkla otizm ve Angelman sendromunun belirtileriyle karışır. Klinik belirtiler arasında baş büyüme hızının azalması ve bazen mikrosefali, küçük el ve ayaklar bulunur. Stereotipik ve yineleyici el hareketleri de gözlenir. Bilişsel bozukluk ve gerileme döneminde de sosyalleşme sorunları da belirtiler arasında görülür. Okula girdikleri dönemde sosyalleşme genellikle düzelir. Rett sendromu olan kız çocuklar gastrointestinal bozukluklara yakalanmaya yatkındır ve %80’i nöbet geçirir. Hemen hemen hiç sözel becerileri yoktur ve kadınların %50’si yürüyemez. Skolyoz, büyüme eksikliği ve kabızlık çok yaygındır ve sorunlu olabilir.
Hunter sendromu ya da Tip II mukopolisakkaridoz, iduronat-2-sülfataz (I2S) enziminin eksikliğinden ya da yokluğundan kaynaklanan lizozomal depo hastalığıdır. Doktor Charles A. Hunter (1873-1955) tarafından ilk kez 1917 yılında tanımlandığından, Hunter sendromu olarak adlandırılır.

Eril (♂), canlı, sperm üreten fizyolojik bir biyolojik cinsiyet. Her sperm hücresi döllenme işlemi sırasında daha büyük bir dişi gameti ile kaynaşabilir. Bir erilin cinsel yolla üremeyi gerçekleştirmesi için dişiye ait en az bir yumurta hücresine ulaşması gerekir, fakat bazı canlılar eşeyli üreyebildikleri gibi eşeysiz olarak da üreyebilir. Eril insanların (erkeklerin) da içinde bulunduğu birçok eril memeli türünde Y kromozomu bulunurken eril kuşlar ve bazı eril sürüngenlerde ise z kromozomu bulunur; bu kromozomlar eril üreme organlarının gelişmesi için gerekli olan daha fazla miktarda testosteronun üretilmesini sağlar.

Genetik hastalıklar , bir ailede kuşaktan kuşağa aktarılabilen patolojileri niteleyen tanımlamadır. Kalıtsal hastalıkların gelecek kuşaklara aktarılmasında etkili olan faktörlerler, genlerdeki ve kromozomlardaki yapısal değişikliklerdir.
XXXY sendromu, bireylerin iki ekstra X kromozomuna sahip olduğu bir cinsiyet kromozomu anöploidi ile karakterize edilen genetik bir durumdur. Çoğu durumda insanlar iki cinsiyet kromozomuna sahiptir: bir X ve bir Y veya iki X kromozomu. İşlevsel bir SRY genine sahip bir Y kromozomunun varlığı, erkekliği belirleyen genlerin ekspresyonuna neden olur. Bu nedenle, XXXY sendromu cinsiyet kimliğinden bağımsız olarak yalnızca biyolojik erkekleri etkiler. XXXY sendromlu erkeklerde ek iki X kromozomu, tipik 46 yerine 48 kromozoma sahip olmalarına neden olur. XXXY sendromu bu nedenle genellikle 48,XXXY olarak adlandırılır. Bilişsel ve davranışsal problemler, taurodontizm ve kısırlık dahil olmak üzere bu sendromla ilişkili çok çeşitli semptomlar vardır. Bu sendrom genellikle ebeveynlerin gametlerinden birinde yeni bir mutasyon yoluyla kalıtılır, çünkü bundan etkilenenler genellikle kısırdır. XXXY'nin her 50.000 erkek doğumdan birini etkilediği tahmin edilmektedir.
Anorşi, bir cinsiyet gelişim bozukluğu olup XY karyotipe sahip bir bireyin ki genelde eril cinsiyeti belirtir, doğuştan testislerinin olmamasına denir. Döllenme'yi takip eden birkaç hafta içerisinde embriyo ilkel gonadları geliştirir ve bu yapılar üreme sisteminin gelişimini sağlayan hormonları üretir. Eğer testisler sekiz hafta içinde gelişmezse, bebekte dişi genitalya gelişir. Eğer testiler gelişmeye başlar ancak 8 ila 10 hafta arasında kaybedilir veya fonksiyon göstermezse bebekte kuşkulu genitalya oluşur. Bununla birlikte, eğer testisler 14 haftadan sonra kaybedilirse, bebekte kısmi eril genitalya ile birlikte gonadların belirgin yokluğu görülür.