Mutasyon ya da değişinim, bir canlının genomu içindeki DNA ya da RNA diziliminde meydana gelen kalıcı değişmelerdir. Mutasyona sahip bir organizma ise mutant olarak adlandırılır.
Kromozom 2, 22 çift insan otozomal kromozomlarından genellikle ikinci en büyük olanıdır. 242 milyondan fazla baz çiftinden oluşan ve toplam DNA'nın %8'ini kapsayan bir kromozomdur. İnsanlarda normalde bir çift olarak bulunur. Kromozom 2, insan ve diğer maymunların ortak kökenli olması lehine çok güçlü kanıtlar sunmaktadır.
Y kromozomu, iki eşey kromozomundan biridir. Bütün erkeklerde normalde "44, XY" normal karyotip olarak X kromozomuya birlikte bulunur.
Kromozom anomalileri; bir kromozomda meydana gelen yapısal ya da sayısal değişiklikleri gösterir. Genellikle mayoz ve mitozu izleyen hücre bölünmesi sırasında meydana gelen hatalardan kaynaklanırlar. Birkaç farklı türü bulunmakla beraber, genel olarak sayısal ve yapısal anomaliler olarak iki ana gruba ayrılırlar.
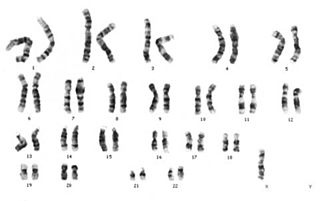
Turner sendromu, kromozom anomalisi sonucu ortaya çıkan ve kız çocuklarını etkileyen bir sendromdur; bir dişide eşey kromozomlarından birinin bulunmaması sonucu ortaya çıkar. Turner sendromluların fenotipi dişi olarak görülür fakat; eşey organları ve eşey hücreleri gelişmez. Kısır bireylerdir.
XYY sendromu veya süper erkek sendromu; erkek bireyin iki Y kromozomu taşımasına neden olan bir eşey kromozomlarında meydana gelen anöplodi durumudur.

Translokasyon, bir kromozomun kaybolan parçasının ya da kopan bir parçasının başka bir kromozoma yapışması şeklinde görülen kromozom anomalilerindendir.
Akraba evliliği ; genetik hastalıkların epidemiyolojisini etkileyen önemli etmenlerden biridir ve dünya toplumunun en az %20'si tarafından yeğlenmektedir. Doğan çocukların en azından %8,4'ü akraba evliliklerinden doğmaktadır.

Frajil X sendromu, X kromozomuyla ilişkili bir zeka (mental) gerilik sendromudur.

TLR7 ya da Toll benzeri reseptör 7, bağışıklık sisteminde rol oynayan Toll benzeri reseptörler ailesinin bir üyesi olan bir gen ve bunun ürünü olan proteindir.
Genetik bozukluk, genlerde ve kromozomlarda görülen anomaliler sonucu ortaya çıkan durumdur. Kanser gibi bazı hastalıklar yaşam sırasında edinilen ve bazı hücrelerde görülen genetik anomaliler nedeniyle olsa da "genetik hastalık" terimi genellikle vücuttaki tüm hücrelerde bulunan ve döllenmeden beri var olan hastalıklar için kullanılır. Bazı genetik bozukluklar, sperm ve yumurtalar gibi üreme hücrelerini oluşturan mayoz bölünme sırasında oluşan kromozom anomalileri nedeniyle ortaya çıkar. Bunlara örnek olarak Down sendromu, Turner sendromu (45X0) ve Klinefelter sendromu sayılabilir. Diğer genetik değişiklikler ebeveynler tarafından tohum hücrelerin oluşturulması sırasında ortaya çıkabilir. Bunlara bir örnek frajil X sendromu ya da Huntington hastalığına neden olabilen üçlü yayılma tekrar mutasyonudur. Hatalı genler ebeveynlerden olduğu gibi alınmış da olabilir. Bu genellikle sağlıklı ama resesif gen taşıyan iki kişinin üremesi ya da hatalı genin dominant olması sonucunda olabilir.
Rett sendromu, yaygın gelişimsel bozukluklardan birisi olarak sınıflandırılan beyinsel gelişim bozukluğudur. Ancak bunun yanlış bir sınıflandırma olduğunu ve benzer şekilde otistik belirtiler gösteren frajil X sendromu, tüberoz skleroz ya da Down sendromunun yaygın gelişimsel bozukluklar olarak sınıflandırılabileceğini önesüren görüşler bulunmaktadır. Bu sendromun belirtileri kolaylıkla otizm ve Angelman sendromunun belirtileriyle karışır. Klinik belirtiler arasında baş büyüme hızının azalması ve bazen mikrosefali, küçük el ve ayaklar bulunur. Stereotipik ve yineleyici el hareketleri de gözlenir. Bilişsel bozukluk ve gerileme döneminde de sosyalleşme sorunları da belirtiler arasında görülür. Okula girdikleri dönemde sosyalleşme genellikle düzelir. Rett sendromu olan kız çocuklar gastrointestinal bozukluklara yakalanmaya yatkındır ve %80’i nöbet geçirir. Hemen hemen hiç sözel becerileri yoktur ve kadınların %50’si yürüyemez. Skolyoz, büyüme eksikliği ve kabızlık çok yaygındır ve sorunlu olabilir.

Glutamat, glutamik asidin anyonudur ve sinirbilimde nörotransmitter olarak görev alır; bir sinir hücresinin başka hücrelere sinyal olarak gönderdiği kimyasallardan biridir. Omurgalı sinir sistemi içerisinde geniş farkla en fazla bulunan nörotransmitterdir. Omurgalı beyninde tüm uyarıcı fonksiyonlarda kullanılır, bu insan beynindeki sinaptik bağlantıların %90'ından fazlasına denk gelir. Bazı beyin bölgelerinde birincil nörotransmitterdir.

Gen duplikasyonu, içinde bir gen bulunan bir DNA bölgesinin herhangi şekilde ikilenmesidir; homolog rekombinasyon sırasında bir hata sonucu, retrotranspozisyon olayı veya tüm bir kromozomun ikilenmesi sonucu meydana gelebilir. Genin kopyası selektif baskıdan yoksun olduğu için, ondaki mutasyonların organizma üzerinde zararlı etkisi olmaz. Dolayısıyla, organizmanın nesilleri boyunca, işlevsel tek kopyalı bir gene kıyasla daha hızlı mutasyona uğrar.
Anti Müller hormon, testislerin sertoli hücreleri tarafından sentezlenen bir hormondur.
Hunter sendromu ya da Tip II mukopolisakkaridoz, iduronat-2-sülfataz (I2S) enziminin eksikliğinden ya da yokluğundan kaynaklanan lizozomal depo hastalığıdır. Doktor Charles A. Hunter (1873-1955) tarafından ilk kez 1917 yılında tanımlandığından, Hunter sendromu olarak adlandırılır.
HUMARA Assay bir tümörün klonal kökenini bulmak için en sık kullanılan yöntemlerden biridir. Metod, X kromozomu inaktivasyonuna dayanmaktadır ve X kromozomu üzerinde bulunan HUMARA geni allellerinin farklı metillenme durumlarına sahip olmaları gerçeğini kullanmaktadır. Bir hücrede X kromozomlarından biri bir kez metillendikten sonra, bu hücreden bölünmeyle oluşacak tüm diğer hücreler aynı X kromozomunu metiller. Yani, aynı ata hücreye sahip (monoklonal) hücreler aynı X kromozomunu inaktive etmişlerdir. HUMARA geninin sahip olduğu üç özellik, onu klonal köken belirleme amacı için oldukça uygun hale getirmiştir:

XXYY sendromu, erkeklerin fazladan bir X ve Y kromozomlarının bulunduğu eşey kromozomları bozukluğu. İnsan hücreleri genellikle anne ve babadan olmak üzere iki cinsiyet kromozomu içerir. Genellikle dişiler iki X kromozomuna (XX), erkekler bir X bir Y kromozomuna (XY) sahiptir. Düzgün çalışan bir SRY geni en az bir Y kromozomunun ortaya çıkmasıyla erkek olur. Bu nedenle XXYY kromozomuna sahip olan insanlar normalde erkek olur. XXYY sendromlu erkekler 46 yerine 48 kromozoma sahiptir. Bu yüzden XXYY sendromu bazen 48, XXYY sendromu olarak yazılır. Yaklaşık her 18.000-40.000 erkek doğumunda bir XXYY sendromlu bireyin doğduğu tahmin edilmektedir.

Genetik hastalıklar , bir ailede kuşaktan kuşağa aktarılabilen patolojileri niteleyen tanımlamadır. Kalıtsal hastalıkların gelecek kuşaklara aktarılmasında etkili olan faktörlerler, genlerdeki ve kromozomlardaki yapısal değişikliklerdir.

Triple X sendromu veya Süper dişi sendromu; X kromozomları ayrılmamış yumurta ile X kromozomu taşıyan normal bir spermin döllenmesi sonucu oluşur. Bu hastalığın tanısı hamilelik döneminde oldukça zordur. Bu bireylerin boy uzunluğu normal bir dişiden uzundur. Öğrenme güçlüğü, zekâ geriliği ve kısırlık görülür. Hastalık her 1000 dişi bebekten 1'inde görülür. 3. X Kromozomu dişinin karyotipindeki normal dişi gelişimini sağlayan genetik hassasiyeti bozar. Zira normalde dişilerdeki iki X kromozomundan 1'i inaktifken bu bireylerde iki X kromozomu inaktiftir. 2n= 44+XXX olarak da ifade edilebilir.