
Şizofreni, benzer belirtilere sahip birtakım ruhsal hastalıklardır.

Obezite, biriken fazla vücut yağının sağlık üzerinde olumsuz bir etkisi olabilecek seviyede çok olması nedeniyle oluşan tıbbi bir durumdur. Bir kişinin ağırlığının kişinin boyunun karesine bölünmesiyle elde edilen bir ölçüm olan Vücut kütle indeksinde (VKİ) genel olarak indeksi 25 kg/m2 ila 30 kg/m2 ve üzeri olanlar obez olarak kabul edilirler. Bazı Doğu Asya ülkelerinde ise daha düşük değerler kullanılmaktadır. Obezite özellikle kalp rahatsızlığı, tip 2 diyabet, obstrüktif uyku apnesi, belirli kanser türleri ve osteoartrit gibi çeşitli hastalıkların olasılığını artırır.
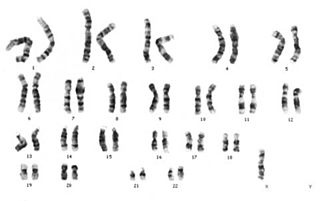
Turner sendromu, kromozom anomalisi sonucu ortaya çıkan ve kız çocuklarını etkileyen bir sendromdur; bir dişide eşey kromozomlarından birinin bulunmaması sonucu ortaya çıkar. Turner sendromluların fenotipi dişi olarak görülür fakat; eşey organları ve eşey hücreleri gelişmez. Kısır bireylerdir.

Diabet ya da Diabetes mellitus, sıklıkla yalnızca diabet ya da diyabet veya halk arasında şeker hastalığı olarak adlandırılan, genellikle kalıtımsal ve çevresel etkenlerin birleşimi ile oluşan ve kandaki glukoz seviyesinin aşırı derecede yükselmesiyle (hiperglisemi) sonuçlanan metabolik bir bozukluktur. Vücutta kan şekerinin düzenlenmesi pek çok sayıda kimyasal madde ve hormonun karmaşık etkileşimi sonucunda sağlanır. Şeker metabolizmasının düzenlenmesinde rol oynayan hormonlardan en önemlisi pankreasın beta hücrelerinden salgılanan insülin hormonudur. Diyabetes Mellitus ya insülin salgılanmasındaki yetersizlik ya da insülinin etkisindeki veya insülin cevabındaki bir bozukluk sonucunda ortaya çıkan yüksek kan şekerinin yol açtığı birkaç grup hastalığı tanımlamak için kullanılan ortak bir terimdir.
Hunter sendromu ya da Tip II mukopolisakkaridoz, iduronat-2-sülfataz (I2S) enziminin eksikliğinden ya da yokluğundan kaynaklanan lizozomal depo hastalığıdır. Doktor Charles A. Hunter (1873-1955) tarafından ilk kez 1917 yılında tanımlandığından, Hunter sendromu olarak adlandırılır.
Konjenital bozukluk olarak da bilinen doğum kusuru, nedeni ne olursa olsun doğumda mevcut olan anormal bir durumdur. Doğum kusurları fiziksel, zihinsel veya gelişimsel engelliliklerle sonuçlanabilir.
Tip 2 diabetes mellitus önceki adıyla insüline bağımlı olmayan diyabet (NIDDM) veya erişkin dönemde ortaya çıkan diyabet –, insülin direnci ve buna bağlı insülin eksikliği bağlamında yüksek kan şekeri ile karakterize edilen bir metabolik bozukluktur. Bu, pankreastaki adacık hücrelerinin yok oluşundan kaynaklanan kesin bir insülin eksikliği bulunan tip 1 diyabetin tam tersine bir durumdur. Klasik semptomlar arasında aşırı susama, sık idrara çıkma ve sürekli açlık bulunmaktadır. Diyabet vakalarının %90’ı tip 2 diyabetten oluşurken tip 1 diyabet ile gestasyonel diyabet, geri kalan %10’unu oluşturur. Genetik olarak obeziteye yatkın olan insanlarda tip 2 diyabetin ana sebebinin obezite olduğu düşünülmektedir.
Williams sendromu , 7. kromozomun uzun kolunda 26 genin silinmesiyle ortaya çıkan; ektodermal displazi bulguları da içeren, otosomal dominant geçen kalıtsal bir sendromdur. Genel gelişme geriliği izlenir, hastaların çoğu zayıftır. Kafatasındaki gelişme duraklamasının sonucu olarak oldukça geniş bir alın vardır. Yabancılara kolay güvenme, geç gelişen dil becerileri, kalp rahatsızlığı, geç gelişen koordinasyon-denge becerisi gibi sonuçlar doğuran nörolojik bozukluktur. Algılama (kognitif) sorunları vardır, psikiyatrik bulgularla karşılaşılabilir. Ses telleri felci nedeniyle ses kabadır. Hasta aktiftir ve mutlu bir görünüm ile aşırı dostça davranış sergiler. Uyku sorunları ve zeka geriliği olabilir.
Erişkinlerde latent otoimmün diyabet ('LADA), yetişkinlikte ortaya çıkan, genellikle çocuklarda tanı konulan tip 1 diyabetten daha yavaş bir başlangıç seyrinde olan bir tip 1 diabetes mellitus tipidir. LADA'lı yetişkinler başlangıçta yaşlarına göre özellikle de güçlü bir aile öyküsü veya obezite gibi tip 2 diyabet için risk faktörleri varsa tip 2 diyabete sahip olarak yanlış teşhis edilebilirler.
Prediyabet, diyabet teşhisi için gerekli olan tüm semptomların bulunmadığı ancak kan şekerinin anormal derecede yüksek olduğu diyabetes mellitusun ön safhasıdır. Bu aşamaya genellikle "gri alan" denir. Bir hastalık değildir; Amerikan Diyabet Derneği'ne göre; "Prediyabet kendi başına klinik bir durum olarak görülmemeli, daha çok diyabet ve kardiyovasküler hastalık (KVH) için bir risk faktörü olarak görülmelidir". Prediyabet obezite, yüksek trigliserit ve/veya düşük HDL kolesterol şeklindeki dislipidemi ve hipertansiyon ile ilişkilidir. Bu nedenle metabolik bir diyatezi veya sendromdur ve genellikle belirti (semptom) vermez ve verdiği tek semptom yüksek kan şekeridir.

Diyabetes insipitus (DI) , yüksek miktarda seyreltik idrar ve aşırı susuzluk hissi ile karakterize bir durumdur. Üretilen idrar miktarı günde yaklaşık 20 litre kadar olabilir. Sıvı alımının azaltılması idrarın konsantrasyonu üzerinde çok az etkiye sahiptir. Komplikasyonlar dehidratasyon veya nöbetleri içerebilir.
Tip 1 diabetes mellitus, pankreas tarafından ya çok az ya da hiç insülin üretilmeyen bir diyabet şeklidir. Tedavi edilmemesi vücutta yüksek kan şekeri seviyesine neden olur. Klasik belirtiler sık idrara çıkma, susuzluğun artması, açlığın artması ve kilo kaybıdır. Ek belirtiler arasında bulanık görme, yorgun hissetme ve yara iyileşmesinin bozulması olabilir. Belirtiler tipik olarak çok kısa bir süre içinde gelişir.
APECED sendromu, ektodermal displazi bulguları içeren, otosomal dominant ya da otosomal resesif geçen kalıtsal bir sendromdur. İlk belirtiler çocukluk yaşlarında ortaya çıkar. Primer immun yetmezlik sendromlarından biridir.
Marshall-Smith sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Sotos sendromunun fenotipi sayılan Malan sendromuna genetik temelde kardeş (allelik) bir sendrom olarak nitelendiren uzmanlar vardır.

Nöromiyelitis optika (NMO), optik sinir ve omuriliğin (miyelit) enflamasyonu ile karakterize, otoimmün, inflamatuar ve demiyelinizan bir hastalıktır. Devic hastalığı olarak da bilinir. Optik nörit ve miyelit aynı anda veya art arda ortaya çıkabilir. NMO ilk kez Fernan Gault ve Eugene Devic tarafından keşfedilmiştir. 1894 yılında bu ikili NMO'yu akut ve ağır optik nörit ve miyelit olarak tanımlamışlardır. Monofazik olarak düşünülen bu hastalığın, 1996 yılında yineleyici bir seyir gösterebileceği kabul edilmiştir.
Gerstmann-Sträussler-Scheinker sendromu (GSS), 20 ila 60 yaş arasındaki hastaları etkileyen, oldukça nadir görülen, genellikle ailesel, ölümcül nörodejeneratif bir hastalıktır. Özel olarak kalıtsaldır ve tüm dünyada yalnızca birkaç ailede bulunur. Bununla birlikte, insan prion proteini olan PRNP'nin oynadığı nedensel rol nedeniyle bulaşıcı süngerimsi ensefalopatiler (TSE) ile sınıflandırılmıştır. GSS ilk olarak 1936'da Avusturyalı doktorlar Josef Gerstmann, Ernst Sträussler ve Ilya Scheinker tarafından rapor edildi.

Diyabetik böbrek hastalığı olarak da bilinen diyabetik nefropati, diabetes mellituslu kişilerde meydana gelen kronik böbrek fonksiyonu kaybıdır. Diyabetik nefropati, küresel olarak kronik böbrek hastalığının (KBH) ve son evre böbrek hastalığının önde gelen nedenleridir. İdrara sızan protein üçlüsü, hipertansiyonla birlikte kan basıncının yükselmesi ve ardından böbrek fonksiyonlarının düşmesi, birçok KBH formunda ortak olarak görülür. Glomerüllerin hasar görmesi nedeniyle idrardaki protein kaybı büyük olabilir ve nefrotik sendrom olarak adlandırılan genel vücut şişmesi (ödem) ile sonuçlanan düşük serum albüminine neden olabilir. Diyabetik nefropati genellikle yıllar içinde yavaş ilerler.
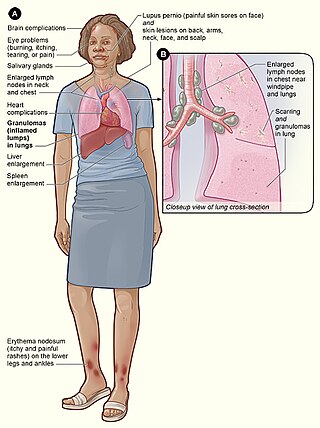
Löfgren sendromu, bir akut sarkoidoz türüdür; bu hastalık, göğüsteki lenf nodlarında büyüme, bacaklarda dokunmaya hassas kırmızı nodüller, ateş ve artritle karakterize bir inflamatuar hastalıktır. Bu hastalık kadınlarda erkeklere kıyasla daha sıktır ve, İskandinavya, İrlanda, Afrika ve Puerto Rika kökenli insanlarda daha sık görülür. Bu hastalık, 1953'te İsveçli bir klinisyen olan Sven Halvar Löfgren tarafından tanımlanmıştır. Bazı kişiler, bu durumun kesin olmayarak tanımlandığını düşünmektedir.
Albinizm-siyah saç teli-hücre göç bozukluğu, bir bireyin fiziksel görünümünü ve fizyolojisini etkileyen durumları tanımlayan terim ve kavramların kısaltmasıdır: (1) A – albinizm, (2) B – siyah saç teli, (3) C – bağırsak nörositlerinin hücre göç bozukluğu ve (4) D – sinirsel tip işitme kaybı. Bu sendrom, endotelin B reseptör geni (EDNRB) mutasyonundan kaynaklanır.
Abruzzo–Erickson sendromu, sağırlık, dışa çıkık kulaklar, kolobom, yarık damak veya damak rugozitesi, radyal sinostoz ve kısa boy ile karakterize son derece nadir görülen bir hastalıktır. İlk olarak 1977 yılında Abruzzo ve Erickson tarafından, iki erkek kardeş, anneleri ve anne amcalarından oluşan bir ailede değişken şekilde ifade edilen bir CHARGE benzeri sendrom olarak tanımlanmıştır. Bu ailenin üyeleri, CHARGE belirtilerinin birçoğunu sergilemiş, ancak koanal atrezi görülmemiş ve erkek kardeşlerde tipik genital gelişim yaşanmıştır. Bu bozukluğun yakın zamanda keşfedilmesi nedeniyle etiyolojisi tam olarak bilinmemektedir, ancak X-kromozomundaki TBX22 genindeki mutasyonlardan kaynaklandığı düşünülmektedir. Hastalık, X'e bağlı resesif bir şekilde kalıtılır. Şu anda bilinen bir tedavisi yoktur, ancak belirtileri tedavi edilebilir.
