Waardenburg sendromu
| Waardenburg sendromu | |
|---|---|
| Diğer adlar | Klein–Waardenburg syndrome (tip 3), Shah–Waardenburg syndrome (tip4) |
 | |
| Tip 1 Wwaardenburg sendromlu bir hastanin yüz görüntüsü (Jan van der Hoeve'nin açıklaması, 1916) | |
| Uzmanlık | Medikal genetik |
Waardenburg sendromu, en azından bir dereceye kadar doğuştan işitme kaybı ve pigmentasyon eksiklikleri ile karakterize edilen, parlak mavi gözleri (veya bir mavi göz ve bir kahverengi gözü ), poliosis veya açık ten lekelerini içerebilen bir grup nadir genetik durumdur. Bu temel özellikler, durumun tip 2'sini oluşturur; tip 1, olarak adlandırılan gözlerin iç köşeler arasında daha geniş bir aralık olan telekantus veya distopia kantorum olarak adlandırılan tipi de mevcuttur.[1] Nadir görülen tip 3'te, kollar ve eller de bozuk, kalıcı parmak kontraktürleri veya kaynaşmış parmaklar, tip 4'te ise kişide bağırsak disfonksiyonuna yol açan doğuştan sinir eksikliği olan Hirschsprung hastalığı vardır.[2][3] Ayrıca, gelişimsel gecikme ve kas tonusu anormallikleri gibi merkezi sinir sistemi semptomlarına neden olabilecek en az iki tip (2E ve PCWH) vardır.[4]
Sendrom, embriyonik gelişim sırasında nöral krest hücrelerinin bölünmesini ve göçünü etkileyen birkaç genden herhangi birinde meydana gelen mutasyonlardan kaynaklanır (dahil olan bazı genler aynı zamanda nöral tüpü de etkiler).[5] Nöral krest hücreleri, nöral tüpün kapatılmasından sonra, vücudun farklı bölgelerinde, melanositler, çeşitli kemikler, yüz, iç kulak kıkırdağı ve periferik bağırsakların sinirleri dahil olmak üzere çeşitli merkezi olmayan sinir sistemi hücreleri oluşturmaya devam eden kök hücrelerdir.[6] Tip 1, PAX3 genindeki bir mutasyondan kaynaklanırken, mutasyona uğradığında en sık tip 2'ye neden olan gen MITF'dir .[1][7] Tip 3, tip 1'in daha şiddetli bir halidir ve aynı gendeki bir mutasyondan kaynaklanırken, tip 4'e çoğunlukla SOX10'daki bir mutasyon neden olur .[2][8] Diğer genlerdeki mutasyonlar da farklı türlere neden olabilir ve bunlardan bazılarına kendi harflerinden alt tür isimleri verilmiştir. Çoğu tip otozomal dominanttır .
Waardenburg sendromunun tahmini yaygınlığı 42.000'de 1'dir.[5][8] Tip 1 ve 2 en yaygın olanlardır ve sırasıyla vakaların yaklaşık yarısını ve üçte birini içerirken, tip 4 vakaların beşinci ve tip 3 %2'sinden daha azını oluşturur. Doğuştan sağır insanların tahminen %2-5'i Waardenburg sendromuna sahiptir. Sendromun tanımları 20. yüzyılın ilk yarısına kadar uzanmaktadır, ancak adını 1951'de tanımlayan Hollandalı göz doktoru ve genetikçi Petrus Johannes Waardenburg'dan almıştır.[9][10] Alt türleri sonraki on yıllarda aşamalı olarak keşfedildi ve çoğunlukla 1990'larda ve 2000'lerde bunlara atfedilen genlere sahipti.
Belirti ve bulgular
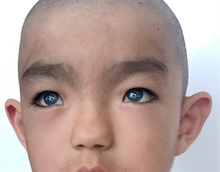
Waardenburg sendromunun, semptomlarda bazı varyasyonlara sahip çok sayıda farklı türü vardır ve semptomlar, aynı tipte olanlar arasında farklılık gösterebilir. Waardenburg sendromunun tüm tiplerinde tutarlı olan iki özellik, bir dereceye kadar doğuştan sensörinöral işitme kaybı ve en tutarlı şekilde gözlerde olmak üzere bir dereceye kadar pigmentasyon eksiklikleridir.[11]
Tip 1
Tip 1, doğuştan sensörinöral işitme kaybı, başın ön merkezinde beyaz bir saç tutamağı (çocuk felci ) gibi saçta pigment eksiklikleri veya erken beyazlaşma, farklı renkli gözler gibi gözlerde pigment eksiklikleri (tam adı heterokromi iridum), bir göz (sektörel heterokromi birden fazla renk) veya parlak mavi göz, cilt depigmentasyon ve gözlerin iç köşeler arasında daha geniş bir aralık boşluk: telekantus veya distopia kantorum. Tip 1 ile ilişkili diğer yüz özellikleri yüksek bir burun köprüsü, bir düz burun ucu, bir de unibrow (synophrys) içerebilir, burun deliklerinde daha küçük kenarlar (alae) ya da bir düz philtrum.[1]
Tip 2
Tip 2'yi tip 1'den ayıran fark, hastaların gözlerin iç köşeleri (telecanthus / distopia canthorum) arasında daha geniş bir açıklığa sahip olmamasıdır. Sensörinöral işitme kaybı, bu tipte daha yaygın ve daha şiddetli olma eğilimindedir.[7][12] Mutasyona uğradığında bu türe neden olan en yaygın gen MITF'dir (tip 2A olarak sınıflandırılır). Bu gende mutasyona sahip iki birey, her iki mutasyonu da taşıyan (homozigot) bir çocuğa sahip olurlarsa ve bunun için %25 olasılık ile, çocukta iriste bir delik (koloboma), küçük gözler (mikroftalmi), sertleşmiş kemikler (osteopetroz), makrosefali, albinizm ve sağırlık gibi ek semptomlar mevcut olacatır.
SNAI2'nin her iki kopyasında da mutasyonlarla tanımlanan iki bilinen hasta vardır (tip 2D olarak sınıflandırılır); bu bireyler Waardenburg sendromu tip 2 ile başvurdu, ancak saç pigmentasyonu eksiklikleri yoktu.[13]
Waardenburg sendromu tip 2'ye SOX10'daki (tip 2E olarak sınıflandırılan) bir mutasyon neden olduğunda, bazı durumlarda birden fazla nörolojik semptomla ortaya çıkabilir. Bunlar, gelişimsel gecikme, erken çocukluk çağı nistagmus, artmış kas tonusu, beyaz cevher anomalileri veya beyinde hipomiyelinizasyon, otistik davranış ve vestibüler sistem veya koklea gibi birçok iç kulak yapısının az gelişmişliğini veya tamamen yokluğunu içerebilir. Beyindeki eksik bir koku soğanı nedeniyle koku alma duyusu eksikliği (anosmi) de mevcut olabilir.[4]
Tip 3
Klein-Waardenburg sendromu veya Waardenburg-Klein sendromu olarak da bilinen tip 3, tip 1 ile aynı semptomlara sahiptir (ve aynı gendeki mutasyonlardan kaynaklanır) ancak kolları ve elleri etkileyen ek semptomları vardır. Bunlar, gelişmemiş kaslar nedeniyle parmakların eklem kontraktürlerini (kamptodaktili) ve ayrıca kaynaşmış parmakları (sindaktili) veya kanatlı kürek kemiğini içerebilir. Mikrosefali ve gelişimsel gecikme de mümkündür.[2]
Tip 4
Shah-Waardenburg sendromu veya Waardenburg-Shah sendromu olarak da bilinen tip 4, tip 2 ile aynı özelliklerin çoğuna sahiptir (yani telecanthus veya belirgin daha geniş göz boşluğu), ancak doğuştan bir eksiklik olan Hirschsprung hastalığının eklenmesi ile bağırsak işlev bozukluğuna yol açan bağırsaklardaki sinirler mevcuttur.[3][14] Ek olarak, işitme kaybı tip 2'deki kadar yaygın değildir. Nadiren, Waardenburg sendromunun bu formunda yarık dudak bildirilmiştir.[15]
Tip 4 ayrıca, tip 4C olarak bilinen SOX10'daki (tip 2E ile aynı gen) bir mutasyondan da kaynaklanabilir; Bu tipte işitme kaybı çok yaygın ve şiddetlidir.[16]
PCWH
Tip 2E ve tip 4C'de yer alan gen olan SOX10'daki bir mutasyon, bazen her iki tipte de semptomlarla sonuçlanabilir (bazen tip 2E'de görüldüğü gibi nörolojik semptomlar ve tip 4'te görüldüğü gibi Hirschsprung hastalığı). Bu olduğunda, periferik demiyelinizan nöropati - merkezi dismiyelinleştirici lökodistrofi - Waardenburg sendromu - Hirschsprung hastalığı (PCWH) olarak adlandırılır.[17][18]
Sebebi

Sınıflandırma tablosu
| Tür | OMIM | Gen | Yer yer | Miras |
|---|---|---|---|---|
| Tip 1 (WS1) | 193500 21 Temmuz 2015 tarihinde Wayback Machine sitesinde arşivlendi. | PAX3 | 2q36.1 [19] | Otozomal dominant |
| Tip 2A (WS2A, orijinal olarak WS2) | 193510 21 Temmuz 2015 tarihinde Wayback Machine sitesinde arşivlendi. | MITF | 3p14.1 – p12.3 | Otozomal dominant |
| Tip 2B (WS2B) | 600193 23 Aralık 2019 tarihinde Wayback Machine sitesinde arşivlendi. | WS2B | 1p21 – p13.3 | Otozomal dominant |
| Tip 2C (WS2C) | 606662 7 Aralık 2019 tarihinde Wayback Machine sitesinde arşivlendi. | WS2C | 8p23 | Otozomal dominant |
| Tip 2D (WS2D) | 608890 21 Temmuz 2015 tarihinde Wayback Machine sitesinde arşivlendi. | SNAI2 | 8q11 | Otozomal resesif |
| Tip 2E (WS2E) | 611584 28 Ocak 2017 tarihinde Wayback Machine sitesinde arşivlendi. | SOX10 | 22q13.1 | Otozomal dominant |
| Tip 3 (WS3) | 148820 9 Mayıs 2013 tarihinde Wayback Machine sitesinde arşivlendi. | PAX3 | 2q36.1 | Otozomal dominant veya otozomal resesif |
| Tip 4A (WS4A) | 277580 28 Ocak 2017 tarihinde Wayback Machine sitesinde arşivlendi. | EDNRB | 13q22 | Otozomal dominant veya otozomal resesif |
| Tip 4B (WS4B) | 613265 28 Kasım 2020 tarihinde Wayback Machine sitesinde arşivlendi. | EDN3 | 20q13 | Otozomal dominant veya otozomal resesif |
| Tip 4C (WS4C) | 613266 28 Ocak 2017 tarihinde Wayback Machine sitesinde arşivlendi. | SOX10 | 22q13.1 | Otozomal dominant |
Tedavi
Şu anda Waardenburg sendromunun tedavisi yoktur. Pratik öneme sahip olma olasılığı en yüksek olan belirti sağırlıktır ve bu, geri döndürülemez başka herhangi bir sağırlık gibi değerlendirilir. Belirgin durumlarda, kozmetik sorunlar olabilir. Sendromla ilişkili diğer anormallikler (nörolojik, yapısal, Hirschsprung hastalığı) semptomatik olarak tedavi edilir.
Toplum ve kültür
Popüler kültür
- Robin Cook'un 2001 romanı Shock, bozukluğa sahip bir karakterden bahseder.[20]
- Peter May'in 2006–2017 kitap serisi The Enzo Files'ın kahramanı Enzo MacLeod'da Waardenburg sendromu vardır. Gözleri farklı renkte ve saçında beyaz bir çizgi var.[21][22]
- Bones'un "The Signs in the Silence" 2011 sezonunun 6. bölümünde, ekip şüpheli katilin Waardenburg sendromuna sahip olduğu bir vakayı çözmelidir.[23]
- 2013 yılında Kimberly McCreight tarafından yazılan Reconstructing Amelia adlı kitap, Waardenburg semptomları olan birkaç karakter içeriyor.[24]
- Karen Rose'un 2014 tarihli Closer Then You Think kitabında, Waardenburg sendromlu üç karakter bulunuyor.[25]
- MJ Mandrake tarafından yazılan 2017 tarihli Murder at the Mayan Temple kitabı Waardenburg sendromlu birkaç karaktere yer veriyor .[26][ ][ ]
- Chandler Baker'ın 2019 romanı The Whisper Network, sendromu bir olay örgüsü olarak kullanıyor.[][ <span title="This claim needs references to reliable sources. (December 2019)">alıntı gerekli</span> ][ <span title="This claim needs references to reliable sources. (December 2019)">alıntı gerekli</span> ]
Önemli insanlar
- Popüler Kanadalı YouTube vloggerı Stef Sanjati, Waardenburg sendromu tip 1'e sahip.[27]
Hayvanlar

Waardenburg sendromu tip 2A (MITF'de bir mutasyon ile) köpeklerde, Fleckvieh sığırlarında, vizonlarda, farelerde ve altın bir hamsterda bulunmuştur.[28] Waardenburg sendromunda görüldüğü gibi koklea ve sakülün dejenerasyonu sağır beyaz kedilerde, dalmaçyalılarda ve diğer köpek ırklarında, beyaz vizonlarda ve farelerde de bulunmuştur.[29]
Ayrıca bakınız
Kaynakça
- ^ a b c "OMIM Entry - # 193500 - WAARDENBURG SYNDROME, TYPE 1; WS1". omim.org. 21 Temmuz 2015 tarihinde kaynağından arşivlendi. Erişim tarihi: 7 Aralık 2019.
- ^ a b c "OMIM Entry - # 148820 - WAARDENBURG SYNDROME, TYPE 3; WS3". omim.org. 9 Mayıs 2013 tarihinde kaynağından arşivlendi. Erişim tarihi: 7 Aralık 2019.
- ^ a b "OMIM Entry - # 277580 - WAARDENBURG SYNDROME, TYPE 4A; WS4A". omim.org. 28 Ocak 2017 tarihinde kaynağından arşivlendi. Erişim tarihi: 7 Aralık 2019.
- ^ a b "OMIM Entry - # 611584 - WAARDENBURG SYNDROME, TYPE 2E; WS2E". omim.org. 28 Ocak 2017 tarihinde kaynağından arşivlendi. Erişim tarihi: 7 Aralık 2019.
- ^ a b Pingault (2010). "Review and update of mutations causing Waardenburg syndrome". Human Mutation (İngilizce). 31 (4): 391-406. doi:10.1002/humu.21211. ISSN 1098-1004. PMID 20127975.
- ^ "Neural Crest Development - Embryology". embryology.med.unsw.edu.au. 28 Ekim 2014 tarihinde kaynağından arşivlendi. Erişim tarihi: 13 Aralık 2019.
- ^ a b "OMIM Entry - # 193510 - WAARDENBURG SYNDROME, TYPE 2A; WS2A". omim.org. 21 Temmuz 2015 tarihinde kaynağından arşivlendi. Erişim tarihi: 7 Aralık 2019.
- ^ a b Song (2016). "Hearing loss in Waardenburg syndrome: a systematic review". Clinical Genetics (İngilizce). 89 (4): 416-425. doi:10.1111/cge.12631. ISSN 1399-0004. PMID 26100139.
- ^ Chandra Mohan (1 Eylül 2018). "Case of Waardenburg Shah syndrome in a family with review of literature". Journal of Otology. 13 (3): 105-110. doi:10.1016/j.joto.2018.05.005. ISSN 1672-2930. PMC 6291636 $2. PMID 30559775.
- ^ Waardenburg PJ (September 1951). "A New Syndrome Combining Developmental Anomalies of the Eyelids, Eyebrows and Noseroot with Pigmentary Anomalies of the Iris and Head Hair and with Congenital Deafness; Dystopia canthi medialis et punctorum lacrimalium lateroversa, Hyperplasia supercilii medialis et radicis nasi, Heterochromia iridum totalis sive partialis, Albinismus circumscriptus (leucismus, poliosis) et Surditas congenita (surdimutitas)". Am. J. Hum. Genet. 3 (3): 195-253. PMC 1716407 $2. PMID 14902764.
- ^ "Waardenburg syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov (İngilizce). 28 Ocak 2017 tarihinde kaynağından arşivlendi. Erişim tarihi: 17 Nisan 2018.
- ^ "Waardenburg syndrome". Genetics Home Reference. October 2012. 31 Mayıs 2010 tarihinde kaynağından arşivlendi.
- ^ "OMIM Entry - # 608890 - WAARDENBURG SYNDROME, TYPE 2D; WS2D". omim.org. 21 Temmuz 2015 tarihinde kaynağından arşivlendi. Erişim tarihi: 7 Aralık 2019.
- ^ "Screening of MITF and SOX10 regulatory regions in Waardenburg syndrome type 2". PLOS ONE. 7 (7): e41927. 2012. doi:10.1371/journal.pone.0041927. PMC 3407046 $2. PMID 22848661.
- ^ Pierpont (March 1995). "A family with unusual Waardenburg syndrome type I (WSI), cleft lip (palate), and Hirschsprung disease is not linked to PAX 3". Clinical Genetics. 47 (3): 139-143. doi:10.1111/j.1399-0004.1995.tb03946.x. ISSN 0009-9163. PMID 7634536.
- ^ "OMIM Entry - # 613266 - WAARDENBURG SYNDROME, TYPE 4C; WS4C". omim.org. 28 Ocak 2017 tarihinde kaynağından arşivlendi. Erişim tarihi: 7 Aralık 2019.
- ^ "Orphanet: Search a disease". www.orpha.net (İngilizce). 24 Mart 2008 tarihinde kaynağından arşivlendi. Erişim tarihi: 10 Aralık 2019.
- ^ Verheij (January 2006). "Shah-Waardenburg syndrome and PCWH associated with SOX10 mutations: a case report and review of the literature". European Journal of Paediatric Neurology. 10 (1): 11-17. doi:10.1016/j.ejpn.2005.10.004. ISSN 1090-3798. PMID 16504559.
- ^ "Isolation of two isoforms of the PAX3 gene transcripts and their tissue-specific alternative expression in human adult tissues". Hum. Genet. 93 (3): 270-4. March 1994. doi:10.1007/bf00212021. PMID 7545913.
- ^ Shock (İngilizce). Pan Macmillan. 12 Aralık 2011. s. 175. ISBN 978-1-4472-1796-1. 27 Ağustos 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 23 Aralık 2020.
- ^ Extraordinary People: Enzo Macleod 1 (İngilizce). Quercus. 30 Mayıs 2013. s. 32. ISBN 978-1-78206-885-3. 27 Ağustos 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 23 Aralık 2020.
- ^ Blacklight Blue: Enzo Macleod 3 (İngilizce). Quercus. 13 Haziran 2013. s. 76. ISBN 978-1-78206-887-7. 27 Ağustos 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 23 Aralık 2020.
- ^ "Bones Recap 6.21 "The Signs in the Silence" – Persephone Magazine" (İngilizce). 11 Ekim 2011 tarihinde kaynağından arşivlendi. Erişim tarihi: 14 Aralık 2019.
- ^ Reconstructing Amelia (İngilizce). Simon and Schuster. 1 Nisan 2013. ISBN 978-1-4711-2944-5. 27 Ağustos 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 23 Aralık 2020.
- ^ Closer Than You Think (The Cincinnati Series Book 1) (İngilizce). Headline. 6 Kasım 2014. ISBN 978-0-7553-8999-5. 27 Ağustos 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 23 Aralık 2020.
- ^ "Meet a nice sleuth". Amazon.com. 1 Ocak 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 14 Aralık 2019.
- ^ Edwards (2018). "'I get you're transgender, but what's up with your face?'". BBC News (İngilizce). 28 Ocak 2018 tarihinde kaynağından arşivlendi. Erişim tarihi: 28 Ocak 2018.
- ^ MARKAKIS (1 Ağustos 2014). "Association of MITF gene with hearing and pigmentation phenotype in Hedlund white American mink (Neovison vison)". Journal of Genetics (İngilizce). 93 (2): 477-481. doi:10.1007/s12041-014-0370-3. ISSN 0973-7731. PMID 25189243.
- ^ Strain (2015). "The Genetics of Deafness in Domestic Animals". Frontiers in Veterinary Science (İngilizce). 2: 29. doi:10.3389/fvets.2015.00029. ISSN 2297-1769. PMC 4672198 $2. PMID 26664958.








