Tümör (ur; neoplasm; tumor) tanımı önceleri vücuttaki herhangi bir şişlik ya da kitle için kullanılırdı. Sonraları hücrelerin kuralsız ve sınırsız çoğalmaları nedeniyle oluşan kitleler için kullanılmaya başlandı. Yaşamın herhangi bir döneminde organizmanın bir bölümündeki hücreler biyolojik niteliklerini düzenleyici kurallara uyum göstermez ve sınırsız olarak çoğalabilir (otonomi). Bu nitelikleri içeren bir kitleye tümör ya da neoplazm (neoplasm; yeni gelişen kitle) adı verilir. Tümör kitleleri vücudun kendi hücrelerinden yapılıdır.

Kanserler (Habis tümörler, Malign tümörler), genellikle sürekli ve hızlı büyüyen tümörlerdir. Kapsülleri yoktur, büyürken sınır tanımazlar, çevresindeki dokuların ve damarların içine girerler (invazyon, infiltratif büyüme). Sıklıkla metastaz yaparlar. Tedavi edilmeyen ya da tedavisi gecikmiş kanserler ölümcüldür.

Hemangioma, kan damarlarının iyi huylu tümörüdür. Olguların çoğu baş-boyun bölgesinde, 1/3'ü karaciğerdedir. Çocuklarda görülen iyi huylu tümörlerin yaklaşık %7'sini oluşturan hemangiomalar, puberteyle birlikte kendiliğinden gerilemekte ve silinmektedir. Cerrahi yöntemlerle çıkarılan oluşumlarda yineleme (residiv) olasılığı vardır.

Kromozom 3; 22 çift otozomal insan kromozomlarından 3. olanıdır. İnsanlarda normalde bir çift halinde bulunur. 200 milyon baz çiftine ve toplam hücre DNA'sının %6,5'ine sahiptir. Kromozom 3 muhtemelen 1,100 ile 1,500 arasında gen içermektedir.
Paraneoplastik sendrom bir tümör veya tümörün metastazları ile doğrudan ilgili olmayan, yerleşim yerlerinden uzaktaki, ancak tümörün varlığına bağlı olan ve dolayısı ile tümörün çıkarılmasından sonra gerileyebilen belirti ve bulgularıdır.
Hermansky-Pudlak sendromu, otosomal resessif aktarılan kalıtsal bir tablodur. 10 fenotipi vardır.
Ağız-Yüz-Parmak sendromu tip 1 , ektodermal displazi bulguları da içeren, X-kromozomu aracılığıyla dominant (XLD) geçen kalıtsal bir sendromdur. Simpson-Golabi-Behmel sendromu tip 2 ile alelik bağı olduğu belirlenmiştir. Kız çocuklarında görece sıktır. Erkek fetüsler, kalp ve beyin anomalilerinin neden olduğu intauterin ölümler nedeniyle kaybedilirler. Belirgin bir genel gelişme geriliği saptanır.

Joubert sendromu, kalıtsal bir sendromdur; 36 fenotipinden 34'ü otosomal resesif yolla aktarılır, olguların bir bölümü Meckel sendromu ile çakışmaktadır. Joubert sendromunun tüm fenotiplerinde 3 temel bulgu vardır: santral sinir sistemi anomalileri, fizik gelişme geriliği, solunum sorunları. Serebellum (beyincik) vermisi ve beyin sapı hipoplazisi ile beyin radyolojisinde saptanan “azıdişi bulgusu ” santral sinir sistemi anomalilerinin başında gelir. Ataksi ve hipotoni sık görülür. Bazı hastalarda oksipital meningosel ya da meningomyelosel olabilir. Schinzel acrocallosal sendromu, önemli fenotiplerinden biridir.
Multipl endokrin neoplazi (MEN), endokrin bezlerden kökenli çok sayıda tümörlerinin saptandığı, otosomal dominant yolla aktarılan kalıtsal bir sendrom kümesidir. Endokrin tümörlerin yanı sıra, endokrin nitelik taşımayan organlardan ve dokulardan kökenli tümörler de görülmektedir; endokrin kökenli olsun ya da olmasın, iyi huylu ya da kötü huylu (kanser) tümör özelliklerini taşırlar.

Bardet-Biedl sendromu, hipofiz ve hipotalamus kökenli endokrin sistem sendromlarından biridir. Kalıtsaldır. 24 fenotipi vardır, bunların yalnızca 3’ü önemlidir ve otosomal dominant ya da otosomal resesif yolla aktarılır.

Cowden sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. PTEN hamartoma tümör sendromları kümesi üyelerindendir. Bazı kaynakların listelerinde, PTEN kümesi sendromlarının tümü Cowden sendromu başlığı altında toplanmaktadır.
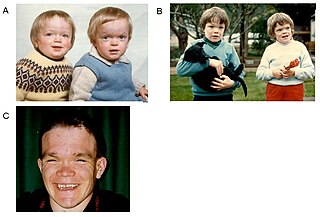
Mannosidozis (mannosidosis), otosomal resesif yolla aktarılan, mannosidaze enzimi eksikliğiyle karakterize kalıtsal bir lizozomal depo hastalığıdır. Alfa-B ve Beta-A olarak nitelendirilen 2 tipi vardır. Alfa-B tipi olgulardaki bulgular çok sayıdadır ve daha güçlüdür.

Meckel-Gruber sendromu, çok sayıda genin etkilenmesinden kaynaklanan, bu nedenle 15 fenotipi olan, otosomal resesif yolla aktarılan, siliyopatiler grubundan, kalıtsal bir sendromdur. Kistik böbrekler, beyin anomalileri ve ensefalosel, polidaktili ve karaciğer anomalileri başlıca bulgulardır. En sık görülen fenotip Meckel sendromu 1, öteki fenotiplerdeki bulguların çok büyük bir bölümünü içerir. Bazı fenotipleri Joubert sendromuyla çakışan nitelikleri içermektedir.

Simpson-Golabi-Behmel sendromu, X-kromozomu aracılığıyla resesif olarak (XLR) aktarılan kalıtsal bir sendromdur; 2 fenotipi vardır.
BOR sendromu, otosomal dominant aktarılan kalıtsal bir sendromdur.

Rubinstein-Taybi sendromu, otosomal dominant yolla aktarılan, 2 fenotipi olan kalıtsal bir sendromdur. 16. kromozom üzerindeki(16p13.3 delesyon) CREBBP(CREB bağlayıcı protein) gen defektiyle bu hastalık oluşur.
Konjenital (doğumsal) böbrek anomalileri, üriner sistemdeki konjenital anomalilerinin önemli bir bölümüdür. Üriner sistemin konjenital anomalilerindeki kalıtımsal nitelikler tam olarak çözümlenememiştir. Olguların yaklaşık 1/5'i kalıtsaldır ve büyük çoğunluğu otosomal dominant yolla aktarılır; otosomal resesif kalıtım oldukça seyrektir. Çoğu olgudaki nedenin gebelikte yaşanan sorunlar olduğu varsayılmaktadır. Böbrek anomalileri, doğumsal üriner sistem anomalilerinin sıkça karşılaşılan sorunlardan biridir; yaklaşık 10 çocuktan 1'i bu tür bir anomali (kusur) ile doğar. Böbrek anomalilerin çoğu yapısal bozukluklar gösterirken bir bölümü de metabolik sorunlarla ortaya çıkar.

Polikistik böbrek hastalığı, böbreklerin görece sık karşılaşılan kistik hastalıklarındandır. 2 tip polikistik böbrek hastalığı vardır;
- Otosomal dominant polikistik hastalık
- Otosomal resesif polikistik hastalık
Kavernöz hemangioma, kapiller hemangiomlardakine benzer yerleşim gösterirler; ancak, daha derinde ve daha büyüktür, sınırları belirsizdir. Çevre dokulara olumsuz etkileri nedeniyle cerrahi yolla çıkarılmaları gerekebilir. 1–2 cm çaplarında, morumsu-kırmızı, yumuşak sünger kıvamında tümörlerdir. Yüzde, kollarda ve bacaklarda daha büyük tümörler görülebilir. Çoğu olguda herhangi bir sorun yaşanmaz. Yüz bölgesindeki tümörler estetik açıdan önemlidir. Yüzeye yakın olanlara gelen travmatik etkiler kanamalara neden olur. İç organ tümörüne radyoloji teknikleriyle tanı konur.

William G. Kaelin Jr., Amerikalı Nobel ödüllü bir hekim ve bilim insanıdır. Harvard Üniversitesi ve Dana-Farber Kanser Enstitüsü'nde tıp profesörüdür. Laboratuvarı tümör baskılayıcı proteinler üzerinde çalışmaktadır. Kaelin, 2016 yılında Albert Lasker Temel Tıbbi Araştırma Ödülü ve AACR Prenses Takamatsu Ödülü'nü aldı. Ayrıca 2019'da Peter J. Ratcliffe ve Gregg L. Semenza ile birlikte Nobel Fizyoloji veya Tıp Ödülü'nü kazandı.
