
El, şempanze, maki ve insan gibi primatlarda birden fazla parmağı barındıran, kolun bilekten parmak uçlarına kadar olan bölümünü tanımlar. Pek çok primat elleri sayesinde tutunma ve tırmanma gibi özelliklere sahiptir. Başparmağın diğer parmaklarla karşılıklı iş görmesi, ufak nesneleri ele alabilme yeteneğini sağlar. Bu özellik sayesinde el, alet kullanımı gibi hassas ve karışık işleri görebilir. Primatların beyninde eli temsil eden alan, diğer hayvanlarınkinden çok daha geniştir. Bu yüzden beyindeki bazı bozuklukların ilk belirtilerinden biri de el parmak hareketinin zarar görmesidir.

Ayak, birçok omurgalıda bulunan anatomik bir yapıdır. Ağırlık taşıyan ve hareket etmeye olanak sağlayan bir uzvun terminal kısmıdır. Ayakları olan birçok hayvanda, ayak, bacağın terminal kısmında, genellikle pençeler veya tırnaklar da dahil olmak üzere bir veya daha fazla segment veya kemikten oluşan ayrı bir organdır.

Klasik gitar, klasik müzikte kullanılan gitar ailesinin bir üyesidir. Bağırsak veya naylondan yapılmış telleri olan bu akustik ahşap telli çalgı, metal telleri kullanan akustik ve elektro gitarın öncüsüdür. Klasik gitar on beşinci ve on altıncı yüzyılda İspanyol vihuela ve gittern'den türetilmiştir, daha sonra on yedinci ve on sekizinci yüzyıl Barok gitarına ve daha sonra on dokuzuncu yüzyılın ortalarında modern klasik gitara dönüşmüştür.
Pfeiffer sendromu kafatası kemiklerinin erken birleşmesi (kraniosinostoz) ile karakterize olmuş kafa ve yüz kemiklerinin şeklini etkileyen bir genetik bozukluktur. Pfeiffer Sendromu el ve ayaklardaki kemikleri de etkilemektedir.

Parmak kemikleri veya falanks kemikleri, çoğu omurgalının ellerinde ve ayaklarındaki parmaklarda bulunan kemiklerdir. Primatlarda, el ve ayak başparmaklarında iki falanks, diğer parmaklarda ise üç falanks vardır. Parmak kemikleri uzun kemikler olarak sınıflandırılır.
Greig sendromu (Greig sefalopolisindaktili sendromu) kalıtsal bir sendromdur. Kafatası ve el parmaklarının birbirlerine yapışık olmasıyla karakterizedir (cephalopolysyndactylia).
Oral-Facial-Digital sendrom II (orofaciodigital sendrom 2, Mohr sendromu; Mohr-Claussen sendromu), ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur. Belirgin bir genel gelişme geriliği saptanır.
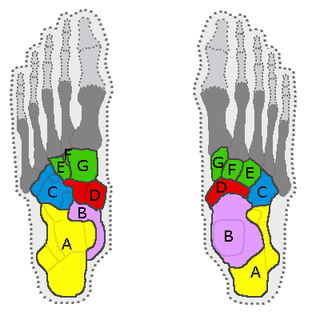
Tarsus, her ayağında tibianın alt ucu ile alt bacağın fibulası ve metatarsus arasında yer alan yedi eklem kemiği kümesi. Orta ayak ve arka ayaktan oluşur.
LADD sendromu; lacrimo-auriculo-dento-digital sendrom; Levy-Hollister sendromu), ektodermal displazi bulguları da içeren, gözyaşı bezi,kulak, diş ve parmak bulgularının baskın olduğu, otosomal dominant yolla geçen kalıtsal bir sendromdur.
OPD sendromu (otopalatodigital sendrom), X-kromozomu aracılığıyla dominant (XLD) yolla geçen kalıtsal bir sendromdur (Gorlin-Cohen sendromu hariç). Kız çocukları daha hafif etkilenir. Tümü benzer bulgular içeren 5 tipi vardır:
- Otopalatodigital sendrom tip I (OPD tip1)
- Otopalatodigital sendrom tip II (OPD tip2)
- Gorlin-Cohen sendromu (ilgili sayfaya gidiniz)
- Melnick-Needles sendromu (tip 1 ile allelik bağlantısı vardır; ilgili sayfaya gidiniz)
- Terminal osseöz displazi ve pigmentli deri defektleri (çok enderdir, ayrıntı verilmemiştir).

Metakarpal kemikler veya tarak kemikleri, insan el iskeletinde parmak kemikleri ile karpal kemikler arasında yer alan kemikler. Metakarpal kemikler ayaktaki metatarsal kemiklere benzerdir.
Femoral-facial sendrom, izole sendromdur; diabetik annelerin bebeklerinde görece sık rastlanır. otosomal dominant yolla aktatılmış az sayıda olgu bildirilmiştir.

Schinzel acrocallosal sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Joubert sendromunun önemli fenotiplerindendir. Santral sinir sisteminde corpus callosum yetersizliği ve zeka geriliği bulguları ile parmaklarda belirgin olan çok sayıda oluşum kusurları saptanır.

Bohring-Opitz sendromu, otosomal dominant yolla aktarılan, gelişme ve zeka geriliği bulgularının ön planda olduğu, hastaların çoğunun çocukluk yaşlarında kaybedildiği bir sendromdur. C sendromu'nun fenotipi olarak benimsenir.
Larsen sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Ana nitelikleri nedeniyle, osteokondrodisplazi ve OPD sendromu 1 ile benzerlikleri vardır. Kalça, dirsek, bilek, diz gibi büyük eklemlerin çıkıkları, ayak deformasyonları, özellikle el başparmaklarda belirgin olan yassılaşma, skolyoz, boyun omurlarının malformasyonlarından kökenli kifoz, spina bifida, iskelet sistemiyle ilgili başlıca bulgulardır. Ayrıca, fibula, iki tarafta da tibianın önündedir (dislokasyon). El ve ayak parmaklarında çeşitli anomalilere rastlanabilir. Tırnaklar kısa ve küçüktür.

Peters’ Plus sendromu , otosomal resesif yolla aktarılan, kalıtsal bir sendromdur. Genel gelişme geriliği nedeniyle boy kısadır. Boyun kalın, alın bombesi yüksektir. Yüz derisinde aşırı kıllanma (hipertrikoz) olabilir. Gözler birbirilerinden uzakçadır (hipertelorizm), göz kapaklarının açıklığı dardır. Görme sorunlarına neden olan Peters anomalisi, glokom, katarakt ve retinafs defektler (koloboma) vardır. İşitme sorunları saptanır; kulaklar küçük ve geridedir, kulak deliği önündeki deride çukurlar görülür. Üst dudak kabarıktır. Yarık dudak ve yarık damak saptanır. Altçene küçüktür (mikrognati). Üst yan kesici dişler eksiktir (hipodonti). Dili ağız tabanına bağlayan bağ kısadır (ankyloglossia).

Asfiksiyan torasik displazi sendromu, otosomal resesif yolla aktarılan, 22 fenotipi olan kalıtsal bir sendromdur. İlk tanımlanan fenotip 1, Jeune sendromu olarak bilinmemektedir. Fenotiplerden ikisi Ellis-van Creveld sendromu kapsamındadır. Saldino-Noonan sendromu, Majewski sendromu, Mainzer-Saldino sendromu ve Beemer-Langer sendromu görece sık rastlanan fenotiplerdir.

Richieri-Costa-Pereira sendromu, çene ve yüz bulgularının yoğun olduğu, iskelet sistemi malformasyonlarının saptandığı, otosomal resesif olarak aktarılan kalıtsal bir sendromdur. Erkek çocuklarında ölü doğum ya da doğumu izleyen günlerde ölümler sık görülür.

Waardenburg sendromu, en azından bir dereceye kadar doğuştan işitme kaybı ve pigmentasyon eksiklikleri ile karakterize edilen, parlak mavi gözleri, poliosis veya açık ten lekelerini içerebilen bir grup nadir genetik durumdur. Bu temel özellikler, durumun tip 2'sini oluşturur; tip 1, olarak adlandırılan gözlerin iç köşeler arasında daha geniş bir aralık olan telekantus veya distopia kantorum olarak adlandırılan tipi de mevcuttur. Nadir görülen tip 3'te, kollar ve eller de bozuk, kalıcı parmak kontraktürleri veya kaynaşmış parmaklar, tip 4'te ise kişide bağırsak disfonksiyonuna yol açan doğuştan sinir eksikliği olan Hirschsprung hastalığı vardır. Ayrıca, gelişimsel gecikme ve kas tonusu anormallikleri gibi merkezi sinir sistemi semptomlarına neden olabilecek en az iki tip vardır.
Werner sendromu (WS), aynı zamanda "yetişkin progeriası" olarak da bilinir, erken yaşlanmanın ortaya çıkmasıyla karakterize, nadir görülen, otozomal resesif geçişli bir genetik hastalıktır.

 . ISSN 1435-232X. PMID 24965254.
. ISSN 1435-232X. PMID 24965254.