Taş adam sendromu
| Taş Adam Sendromu | |
|---|---|
| Diğer adlar | Fibrodisplazi ossifikans progressiva, Munchmeyer hastalığı |
 | |
| Hasar almış yumuşak dokunun kemik olarak geliştiği bir hastalık olan taş adam sendromunun etkileri.(Harry Raymond Eastlack'e ait iskelet) | |
| Olağan başlangıcı | 10 yaşından önce |
| Uzmanlık | Medikal genetik, romatoloji |
| Belirtiler | Devamlı kemik gelişimi |
| Ayırıcı tanı | Fibröz displazi |
| Tedavi | Yok |
| Prognoz | Ortalama yaşam süresi ≈ 40 yıl (eğer iyi bir şekilde idare edilirse) |
| Sıklık | Dünya çapında 801 teyitli vaka (2017); insidansı her bir milyon insanda 0,5’tir. (2 milyonda 1) |
Taş adam sendromu (Munchmeyer hastalığı, FOP, fibrodisplazi ossifikans progressiva veya eski adıyla miyositis ossifikans progressiva);[1] kas, tendon ve ligament gibi fibröz bağ dokularının kemik dokusuna dönüştüğü ve oldukça nadir görülen bir bağ dokusu hastalığıdır. Bir organ sisteminin başka bir organ sistemine dönüştüğü görülen tek tıbbi durumdur.[2] Tedavisi veya çaresi olmayan ciddi ve sakatlayıcı bir hastalıktır.
FOP, ACVR1 geninde ortaya çıkan bir mutasyon sonucu oluşur. Bu mutasyon; kas, tendon ve ligament gibi fibröz dokunun aniden veya travma sonucu oluşan hasar sonucu kemikleşmesine neden olarak vücudun tamir mekanizmasını etkiler. Çoğu vakada, küçük incinmelerde hasar alan kas dokusunun kemik dokuya dönüşmesi sonucu eklemler kalıcı olarak kaynaşabilir. Bu yeni kemiksel oluşum (“heterotopik ossifikasyon” olarak bilinir), eninde sonunda ikincil bir iskelet oluşturacak ve geliştikçe hastanın hareket kabiliyetini sınırlayacaktır. Bu sürecin sonucunda oluşan kemik “normal” kemikle tamamen aynıdır, sadece olmaması gerek yerlerde konumlanmıştır. Dolaylı kanıtlar, hastalığın karakteristik olarak kemik gelişimine sebep olmasının yanı sıra eklem yıkımına da yol açtığını göstermektedir.[3]
Ekstra kemik gelişiminin ameliyatla çıkarılması sonucu vücudun etkilenen bölgeyi ilave kemiklerle “onardığı” görülmüştür. Hastaya bağlı olarak kemik gelişim hızı değişse de yeni kemiklerin kas sisteminin yerini alması ve halihazırda var olan iskelet sistemiyle kaynaşması sonucu bu durum, hastalığa sahip olan kişileri en sonunda hareketsiz bırakır. Bu nedenle FOP’a “taş adam sendromu” lakabı takılmıştır.[4]
Belirtiler
Bilinmeyen nedenlerden dolayı FOP’la doğan çocuklarda sıklıkla kusurlu ayak baş parmağı, eksik bir eklem veya farklı durumlarda küçük eklemlerde göze çarpan bir yumru görülür.[5] FOP oluşumuna neden olan “ateşlenme” genellikle 10 yaşından önce meydana gelir.[5] Fetüslerdeki kemik oluşumuna benzer biçimde ekstra kemik gelişimi, genellikle vücudun üstünden altına doğru ilerler. FOP’a sahip bir çocuk tipik olarak boynundan başlayarak omuzlarına, kollarına ve göğüs bölgesine ilerleyen ve en sonunda ayağa ulaşan ilave kemik gelişimi gösterir.[6]
Spesifik olarak kemikleşme; ilk olarak vücudun dorsal, aksiyal, kranyal ve proksimal bölgelerinde ortaya çıkar. Daha sonra hastalık; ventral, apendiküler, kaudal ve distal bölgelerde ilerler.[5] Ancak, yaralanma sonucu ateşlenmeler nedeniyle hastalık illaki bu sıra ile ilerlemek zorunda değildir. Sıklıkla, hastalığın ateşlenmesini karakterize eden tümör benzeri yumrular aniden ortaya çıkar.
Alevlenmeler sırasında ortaya çıkan kemik gelişimi, çene dahilse ağzı tamamen açamama, sınırlı yemek ve konuşma da dahil, etkilenen eklemlerde hareket kabiliyetinin kaybına sebep olabilir. Spesifik olarak ayak ve ayak bileği eklemlerinde meydana gelen ateşlenmeler de yere düz basma kabiliyetinin sınırlanmasına yol açabilir. Kemik gelişimi ayrıca bireyin yürüme kabiliyetini etkileyecek biçimde kalça veya dizde de ortaya çıkabilir. Göğüs kafesinin çevresinde oluşan ekstra kemik oluşumu, akciğer ve diyaframın genişlemesini engelleyerek solunum sorunlarına yol açar.[5]
Bu hastalık çok nadir olduğundan dolayı kanser veya fibrozis gibi yanlış tanılar konulabilir. Bu durum da hekimlerin biyopsi isteyerek FOP kemiklerinin gelişimini artırmasına neden olabilir.[7] FOP hastalarının kusurlu ayak ve el bai parmakları ile doğması, bozukluğun diğer iskelet problemlerinden ayrılmasını sağlar.[6]
Uygun tıbbi bakım ile ortalama yaşam süresi 40 yıldır. Fakat ertelenmiş tanı, travma ve enfeksiyonlar; beklenen yaşam süresini düşürebilir.[8]
Nedenler
FOP, 2q23-24 kromozomundaki otozomal baskın bir alel sonucu meydana gelir.[9] Bu genin ekspresyonu değişkenlik gösterse de fenotipte kesinlikle görülür. Çoku vaka, gametlerdeki ani mutasyon sonucu ortaya çıkar; FOP’lu hastaların çoğu çocuk sahibi olamaz veya olmamayı tercih eder. Bu duruma benzer fakat daha az feci bir hastalık ise postzigotik bir mutasyon sonucu oluşan fibröz displazidir.
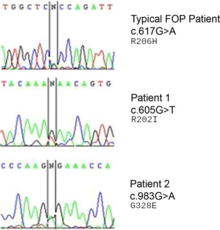
ACVR1 (aktivin benzeri kinaz 2 (ALK2) olarak da bilinir) geninde oluşan bir mutasyon, bu hastalıktan sorumludur.[9] ACVR1, bir BMP tip-1 reseptörü olan aktivin tip-1 reseptörünü kodlar. Mutasyon, kodon 206’nın değiştirilerek ACVR1 proteinindeki arjininin histidine dönüşmesine neden olur.[10][11] Bu değişim, AVCR1’in anormal aktivasyonuna neden olarak bağ ve kas dokusunun sekonder iskelete dönüşmesine yol açar. Bu durum; endotel hücrelerinin ilk olarak mezenkimal kök hücrelerine, sonradan da kemiğe dönüşmesine neden olur.[12] Normalde ACVR1 geni, kondrogenez sinyalizasyonu amacıyla BMP reseptörlerini (Tip I BMPR ve Tip II BMPR) bağlayan aktivin tip-1 transmembran kinaz reseptörlerini kodlar. BMPler, transforme edici büyüme faktörü-beta (TGF-β) olarak bilinen bir protein süperailesine aittir. ACVR1 proteinin BMP reseptörlerine bağlanması, gelişim sırasındaki endokondral kemikleşmenin indüklenmesinin yanı sıra iskelet ve doku homeostazisi için de önemli olan bir sinyal dizisini başlatır.[13]
Genetik
FOP, otozomal baskın bir hastalıktır. Bu sebeple hastalıktan etkilenmiş heterozigot bir ebeveynle beraber etkilenmemiş bir ebeveyne sahip olan bir çocuğun hastalığa sahip olma ihtimali %50’dir. İki hastalıklı birey, hastalıksız bir çocuk sahibi olabilir. Gendeki mutasyon sonucu hastalıksız olan iki ebeveyn de hastalılı bir çocuk dünyaya getirebilir. Hastalığın homozigot formu, heterozigot formundan daha ciddidir.[14]
Kemikleşmeye neden olan protein, genellikle rahimdeyken fetüsün kemikleri oluştuktan sonra inhibe edilir ancak FOP’lu hastaların proteinleri çalışmaya devam eder. FOP’lu hastalarda anormal kemik oluşumu; meydana gelen bir hasar sonucu bağ dokusunun veya kas hücrelerinin apoptoz (hücrenin kendi kendisini öldürmesi) sırasında yanlışlıkla bir kemik onarım enzimini eksprese etmesi ve bu durum sonucu bağışıklık sisteminin yanıtı sırasında lenfositlerin kemik morfogenetik proteini 4‘ü salgılamasıyla gerçekleşir. Sonuç olarak kemik, kendi ayrı iskelet elementlerini oluşturarak normal iskeletten bağımsız olarak gelişir. Ancak bu elementler, normal iskelet kemikleri ile kaynaşabilir.[15] Diyafram, dil, göz dışı kaslarla beraber kalp kası ve düz kaslar da bu işlemlerden ayrı tutulur.[5] Yanlış enzim bağışıklık sisteminin içerisinde yıkılmadan tutulduğu için vücut, yanlış BMP4 içeren lenfositleri yönlendirmeye devam eder. BMP4, normal bir embriyoda iskelet gelişimine yardım eden bir maddedir.[16]
ACVR1 geni, bir kemik morfogenetik protein (BMP) reseptörünü kodlar ve FOP’lu hastalarda bu gen mutasyona uğramıştır. Bu gen, kemik ve kas gelişiminden sorumludur. Tipik mutasyon olan R202H, FKBP1A inhibitörününün GS-döngü aktivasyonuna daha gevşek bağlanmasına neden olur.[17] Bunun sonucunda ACVR1 kapatılmaz ve kemik ile kıkırdağın fazla büyümesiyle beraber eklem kaynaşmaları da görülür.[6] Tipik olmayan mutasyonlar ise benzer şekilde çalışan kalıntılarla ilgilidir. Bazı durumlarda, reseptör kendi aktive edici ligantına bağlanmadan aktive olmuş gibi sinyal yayabilir.[18]
FOP vakalarının çoğu, yeni bir mutasyondan kaynaklıdır: bu insanların ailelerinde bu mutasyona rastlanmaz. Bazı durumlarda ise birey, mutasyonu hastalıklı bir ebeveynden kalıtımla almıştır.[6]
Tanı
Genellikle FOP tanısı radyografilerle konulabilir. Biyopsi gibi gereksiz invaziv prosedürlerden kaçınmak için radyoloji aracılığıyla erken tanı almak oldukça önemlidir. Küçücük veya önemsiz travmalar ya da intramüsküler enjeksiyonlar inflamasyonla hastalığın ilerleyişini hızlandırabildiği için radyoloji çok daha avantajlıdır. Sıklıkla kanser veya enfeksiyonlar gibi diğer iyi huylu oluşumlarla yanlış tanı koyulup hastalığın ilerleyişini kötüleştiren biyopsi alımlarından dolayı doktorlar, bu nadir hastalıktan haberdar olmalıdır.[19]
Hastalığın baş gösterişi, klinik olarak yüksek alkalen fosfataz ve kemiğe özgü alkalen fosfataz değerleri ile ölçülebilir.[20] FOP’un bir diğer göstergesi ise kusurlu birinci distal metatarsal kemik ve noksan veya anormal birinci falanks ya da interfalangeal eklemle beraber kısa baş ayak parmağıdır.[21]
Tedavi
Fibrodisplazi ossifikans progressiva (FOP) için hala bir çare bulunamamasına rağmen onaylanmış bir tedavi yöntemi olan Sohonos’ta (palovaroten) ümit verici bir gelişme görüldü.[22] Önemli bir şekilde; FOP hastasından ameliyatla kemiği çıkarmak, hızlıca yeni bir kemiğin gelişimine neden olabilir.[23] Anestezi etkisi altına giren FOP’lu bireyler; entübasyon ve restriktif akciğer hastalıkları ile ilgili sıkıntılar yaşayabilirken kalbin elektriksel ileti sisteminde de değişiklikler meydana gelebilir.[24] Yumuşak dokuyucu incitebilecek veya bireyi düşürebilecek aktivitelerden kaçınmak gerekir çünkü küçük bir travma bile heterotopik ossifikasyona neden olabilir.[25]
Bu bozukluk için etkili kesin bir tedavi bulunmamasına rağmen kemik hasarı sonucu ortaya çıkan ateşlenmeler ve inflamasyonu baskılayabilecek birtakım anti-inflamasyon ilaçları kullanılabilmektedir. Kesik bölgelerinde hızlı bir kemik gelişimi görülebileceği için FOP hastaları için ameliyat, henüz mümkün değildir. Eklem kasılmalarını serbest bırakmak için yapılan operasyonlar genellikle başarısız olmaktadır ve travma ile ilişkili heterotopik ossifikasyon riskini de artırmaktadır.[5]
Epidemiyoloji
2017 itibarıyla dünya çapındaki toplam vaka sayısının yaklaşık 800 olmasıyla FOP, bilinen en nadir hastalıklardan biri haline gelmiştir.[26] FOP’un tahmini insidansı her bir milyon insanda 0,5 vakadır ve bu hastalık, her etnik kökenden insanı etkileyebilir.[26]
Tarih
FOP’tan etkilenen bireyleri tanımlayan medikal raporlar, 1962’de Dr. Guy Patin’in yazılarına kadar uzanır.[26] FOP, ilk olarak miyositis ossifikans progressiva olarak isimlendirilmiştir ve kemik oluşumuna sebep


olan bir müsküler inflamasyon (miyositis) sonucu meydana geldiği düşünülmüştür.[26] Hastalık, Victor A. McKusick’in 1970’te kas harici başka yumuşak dokuların da (ligamentler gibi) bu bozukluktan etkilenebileceğini bulmasından sonra yeniden adlandırılmıştır.[26]
En çok bilinen FOP vakası, Harry Eastlack (1933-1973) vakasıdır. Durumu, on yaşında ortaya çıkmıştı ve zatürreden Kasım 1973’te ölmeden yani 40. yaşından altı gün önce vücudu tamamen kemikleşmişti ve sadece dudaklarını oynatabilir hale gelmişti. Eastlack, hayatı boyunca başka hiçbir FOP’lu bir hastayla tanışmadı.[27]
Eastlack, vücudunu bilime bağışlamıştır ve iskeleti şu an Philadelphia’daki Mütter Müzesi’ndedir; bu iskelet, FOP ile ilgili çalışmalarda paha biçilemez bir bilgi kaynağı olmuştur. FOP’lu başka birisi olan Carol Orzel de (20 Nisan 1959 - Şubat 2018) vücudunu müzeye bağlamıştır ve iskeleti, Eastlack’inkinin hemen yanına Şubat 2019’da koyulmuştur.[28][29]
Araştırma
İzotretinoin, oral kortikosteroidlerle beraber etidronat ve perheksilin meleat ile yapılan klinik deneyler; hastalığın prevelansının düşüklüğü ve hastalığın farklı seyirleri nedeniyle belirsizlik ortaya çıksa da bu ilaçların etkisiz olduğunu göstermiştir.[20]
Nadir hastalıklara odaklanan birtakım ilaç şirketi, şu anda FOP için farklı tedavi yaklaşımlarına yönelik araştırmaların farklı aşamalarındadır.
Ağustos 2015'te, ABD Gıda ve İlaç Dairesi (FDA) Yetim Ürünler Geliştirme Ofisi, La Jolla Farmasötikal'e FOP için iki yeni bileşik yetim ilaç tanımı verdi. Bu bileşikler, ACVR1’i (ALK2) seçici bir şekilde engellemek için tasarlanmış küçük moleküllü protein kinaz inhibitörleridir.[30]
Ağustos 2015'te Clementia Pharmaceuticals, FOP tedavisi için palovaroteni araştıran faz II klinik deneyine çocukları (6 yaş ve üzeri) kaydetmeye başladı.[31] Klinik öncesi çalışmalar, bir retinoik asit reseptörünün gama agonisti olan palovarotenin, BMP yolundaki ikincil haberci sistemlerin inhibisyonu yoluyla hayvan modellerinde anormal kemik oluşumunu bloke ettiğini göstermiştir.[32] Clementia, daha önce sağlıklı gönüllüler ve kronik obstrüktif akciğer hastalığı olan hastalar da dahil olmak üzere 800'den fazla kişide bileşiği değerlendiren Roche Pharmaceuticals'dan palovaroten lisansını aldı. Palovarotene, FDA'dan Fast Track ünvanını ve hem FDA hem de Avrupa İlaç Ajansı'ndan (EMA) FOP tedavisi için yetim atamalarını aldı.[31]
Eylül 2015'te Regeneron, hastalık mekanizmasına ilişkin ACVR1 reseptörünün aktivin A tarafından aktivasyonunu içeren yeni bilgiler duyurdu. 2016 yılında şirket, sağlıklı gönüllülerde aktivin antikoru REGN


2477 ile ilgili bir faz I çalışması başlattı; 2017 yılında FOP hastalarında faz II denemesi yapıldı.[33]
Bir başka potansiyel terapötik yaklaşım, normal ACVR1 gen ekspresyonunu korurken, bozulma için mutasyona uğramış mRNA'yı hedefleyen alele özgü RNA müdahalesini içerir.[34]
FOP'ta heterotopik kemik oluşumunun mekanizmalarının daha fazla araştırılması, iskelet dışı kemik oluşumunu içeren diğer bozuklukların tedavilerinin geliştirilmesine yardımcı olabilir.
Fibro/adipojenik progenitörler (FAP'ler); ACVR1(R206H) mutasyonuna neden olan FOP taşıyan farelerin hem kaslarında hem de tendonlarında aktivin A'ya bağımlı ektopik kemik oluşumundan sorumlu, hastalığa neden olan hücre tipi olabilir.[35]
Aralık 2019'da Ipsen, büyüme plakalarının erken füzyonuna ilişkin raporlar nedeniyle 14 yaşın altındaki kişiler için kısmi bir klinik durdurma yayınladı.
Son zamanlarda, 2021 itibarıyla, potansiyel bir terapötik aday olan sarakatinib, vahşi tipte ve ACVR1 mutant farelerde güçlü bir heterotopik ossifikasyon inhibitörü olarak faz III klinik deneylerindedir.[36]
Ayrıca bakınız
- Uluslararası FOP Birliği
- Osteogenezis imperfekta, FOP'tan sorumlu genlerdeki mutasyonların neden olduğu, kemiklerin alışılmadık derecede kolay kırılmasıyla karakterize edilen bir durum
- Progresif ossöz heteroplazi
- FOP Arkadaşları
Kaynakça
- ^ "Medical Definition of FIBRODYSPLASIA OSSIFICANS PROGRESSIVA". www.merriam-webster.com (İngilizce). 17 Ekim 2016 tarihinde kaynağından arşivlendi. Erişim tarihi: 27 Eylül 2023.
- ^ Kaplan, Frederick S.; Shen, Qi; Lounev, Vitali; Seemann, Petra; Groppe, Jay; Katagiri, Takenobu; Pignolo, Robert J.; Shore, Eileen M. (2008). "Skeletal metamorphosis in fibrodysplasia ossificans progressiva (FOP)". Journal of bone and mineral metabolism. 26 (6): 521-530. doi:10.1007/s00774-008-0879-8. ISSN 0914-8779. PMC 3620015 $2. PMID 18979151. 23 Temmuz 2023 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ "Here's what happens when your body tissues turn to bone". web.archive.org. 3 Mart 2019. 3 Mart 2019 tarihinde kaynağından arşivlendi. Erişim tarihi: 27 Eylül 2023.
- ^ Verma, Amit Kumar; Aga, Pallavi; Singh, Shailesh Kumar; Singh, Ragini (25 Temmuz 2012). "The stone man disease: fibrodysplasia ossificans progressiva: imaging revisited". BMJ Case Reports. 2012: bcr2012006422. doi:10.1136/bcr-2012-006422. ISSN 1757-790X. PMC 4543882 $2. PMID 22843760. 23 Ocak 2023 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ a b c d e f Kaplan, Frederick S.; Le Merrer, Martine; Glaser, David L.; Pignolo, Robert J.; Goldsby, Robert; Kitterman, Joseph A.; Groppe, Jay; Shore, Eileen M. (Mart 2008). "Fibrodysplasia ossificans progressiva". Best practice & research. Clinical rheumatology. 22 (1): 191-205. doi:10.1016/j.berh.2007.11.007. ISSN 1521-6942. PMC 2424023 $2. PMID 18328989. 16 Aralık 2022 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ a b c d "Fibrodysplasia ossificans progressiva: MedlinePlus Genetics". medlineplus.gov (İngilizce). 28 Temmuz 2018 tarihinde kaynağından arşivlendi. Erişim tarihi: 27 Eylül 2023.
- ^ Obamuyide, HA; Ogunlade, SO (2015). "A tumour for which surgery will do more harm than good: A case report of fibrodysplasia ossificans progressiva". Nigerian Postgraduate Medical Journal (İngilizce). 22 (1): 83. doi:10.4103/1117-1936.163373. ISSN 1117-1936.
- ^ Saleh, Mohammed; Commandeur, Joost; Bocciardi, Renata; Kinabo, Grace; Hamel, Ben (24 Kasım 2015). "Fibrodysplasia ossificans progressiva with minor unilateral hallux anomaly in a sporadic case from Northern Tanzania with the common ACVR1c.617G>A mutation". The Pan African Medical Journal. 22: 299. doi:10.11604/pamj.2015.22.299.8032. ISSN 1937-8688. PMC 4769042 $2. PMID 26966495. 24 Ocak 2023 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ a b Shore, Eileen M.; Xu, Meiqi; Feldman, George J.; Fenstermacher, David A.; Cho, Tae-Joon; Choi, In Ho; Connor, J. Michael; Delai, Patricia; Glaser, David L.; LeMerrer, Martine; Morhart, Rolf (Mayıs 2006). "A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva". Nature Genetics (İngilizce). 38 (5): 525-527. doi:10.1038/ng1783. ISSN 1546-1718. 25 Temmuz 2019 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ "Penn Researchers Discover Gene That Creates Second Skeleton". ScienceDaily (İngilizce). 4 Mart 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 27 Eylül 2023.
- ^ Hatsell, Sarah J.; Idone, Vincent; Wolken, Dana M. Alessi; Huang, Lily; Kim, Hyon J.; Wang, Lili; Wen, Xialing; Nannuru, Kalyan C.; Jimenez, Johanna; Xie, Liqin; Das, Nanditha (2 Eylül 2015). "ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A". Science translational medicine. 7 (303): 303ra137. doi:10.1126/scitranslmed.aac4358. ISSN 1946-6234. PMC 6164166 $2. PMID 26333933. 23 Ocak 2023 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ van Dinther, Maarten; Visser, Nils; de Gorter, David JJ; Doorn, Joyce; Goumans, Marie-José; de Boer, Jan; ten Dijke, Peter (23 Kasım 2009). "ALK2 R206H Mutation Linked to Fibrodysplasia Ossificans Progressiva Confers Constitutive Activity to the BMP Type I Receptor and Sensitizes Mesenchymal Cells to BMP-Induced Osteoblast Differentiation and Bone Formation". Journal of Bone and Mineral Research (İngilizce): 091211115834058-35. doi:10.1359/jbmr.091110. ISSN 0884-0431.
- ^ Lin, Shuxian; Svoboda, Kathy K H; Feng, Jian Q; Jiang, Xinquan (5 Nisan 2016). "The biological function of type I receptors of bone morphogenetic protein in bone". Bone Research. 4: 16005. doi:10.1038/boneres.2016.5. ISSN 2095-4700. PMC 4820739 $2. PMID 27088043.
- ^ Cummings, Michael R. Human Heredity: Principles and Issues Cengage Learning, 2011, [2009]. p. 77 Cummings, Michael R. Human Heredity: Principles and Issues Cengage Learning, 2011, [2009]. p. 77.
- ^ Shore, Eileen M.; Kaplan, Frederick S. (Eylül 2008). "Insights from a Rare Genetic Disorder of Extra-Skeletal Bone Formation, Fibrodysplasia Ossificans Progressiva (FOP)". Bone. 43 (3): 427-433. doi:10.1016/j.bone.2008.05.013. ISSN 8756-3282. PMC 2601573 $2. PMID 18590993.
- ^ Kierszenbaum, Abraham L. (2002). Histology and cell biology: an introduction to pathology. St. Louis: Mosby. ISBN 978-0-323-01639-1. 18 Mart 2023 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ "ACVR1 gene: MedlinePlus Genetics". medlineplus.gov (İngilizce). 8 Haziran 2023 tarihinde kaynağından arşivlendi. Erişim tarihi: 27 Eylül 2023.
- ^ Petrie, Kirsten A.; Lee, Wen Hwa; Bullock, Alex N.; Pointon, Jenny J.; Smith, Roger; Russell, R. Graham G.; Brown, Matthew A.; Wordsworth, B. Paul; Triffitt, James T. (30 Mart 2009). "Novel Mutations in ACVR1 Result in Atypical Features in Two Fibrodysplasia Ossificans Progressiva Patients". PLoS ONE. 4 (3): e5005. doi:10.1371/journal.pone.0005005. ISSN 1932-6203. PMC 2658887 $2. PMID 19330033. 14 Aralık 2022 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ Yamin, Ghiam; Daghighi, Shadi; Mafee, Mahmood (31 Mayıs 2021). "Fibrodysplasia ossificans progressiva (FOP) presenting as a rapidly growing non-calcified neck mass". Journal of Radiology Case Reports. 15 (5): 10-16. doi:10.3941/jrcr.v15i5.4103. ISSN 1943-0922. PMC 8253151 $2. PMID 34276874. 25 Nisan 2022 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ a b Kitoh, Hiroshi; Achiwa, Masataka; Kaneko, Hiroshi; Mishima, Kenichi; Matsushita, Masaki; Kadono, Izumi; Horowitz, John D; Sallustio, Benedetta C; Ohno, Kinji; Ishiguro, Naoki (16 Ekim 2013). "Perhexiline maleate in the treatment of fibrodysplasia ossificans progressiva: an open-labeled clinical trial". Orphanet Journal of Rare Diseases. 8: 163. doi:10.1186/1750-1172-8-163. ISSN 1750-1172. PMC 4015865 $2. PMID 24131551. 23 Ocak 2023 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ "Fibrodysplasia Ossificans Progressiva - Symptoms, Causes, Treatment | NORD". rarediseases.org (İngilizce). 25 Temmuz 2019 tarihinde kaynağından arşivlendi. Erişim tarihi: 28 Eylül 2023.
- ^ Research, Center for Drug Evaluation and (17 Ağustos 2023). "FDA approves first treatment for Fibrodysplasia Ossificans Progressiva". FDA (İngilizce). 25 Ağustos 2023 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ "Fibrodysplasia Ossificans Progressiva (FOP) - OrthoInfo - AAOS". web.archive.org. 21 Haziran 2012. 21 Haziran 2012 tarihinde kaynağından arşivlendi. Erişim tarihi: 28 Eylül 2023.
- ^ NEWTON, M.C.; ALLEN, P.W.; RYAN, D.C. (Şubat 1990). "FIBRODYSPLASIA OSSIFICANS PROGRESSIVA". British Journal of Anaesthesia. 64 (2): 246-250. doi:10.1093/bja/64.2.246. ISSN 0007-0912.
- ^ "The Medical Management of Fibrodysplasia Ossificans Progressiva: Current Treatment Considerations" (PDF). International Fibrodysplasia Ossificans Progressiva Association. January 2020.
- ^ a b c d e f g Martelli, Anderson; Santos, Arnaldo Rodrigues (31 Ekim 2014). "Cellular and morphological aspects of fibrodysplasia ossificans progressiva". Organogenesis. 10 (3): 303-311. doi:10.4161/org.29206. ISSN 1547-6278. PMC 4750545 $2. PMID 25482313.
- ^ Kaplan, Frederick S. (1 Ekim 2013). "The skeleton in the closet". Gene. Historical Medical Genetics: Skeletal Disorders. 528 (1): 7-11. doi:10.1016/j.gene.2013.06.022. ISSN 0378-1119. PMC 4586120 $2. PMID 23810943.
- ^ McCullough, Marie (28 Şubat 2019). "Mutter Museum exhibit grants final wish for woman who turned to bone". www.inquirer.com (İngilizce). 20 Eylül 2023 tarihinde kaynağından arşivlendi. Erişim tarihi: 28 Eylül 2023.
- ^ "Wayback Machine". web.archive.org. 28 Ağustos 2021. 28 Ağustos 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ a b "La Jolla Pharmaceutical Company Receives Orphan Drug Designation for Two Novel Compounds for Fibrodysplasia Ossificans Progressiva". www.businesswire.com (İngilizce). 18 Ağustos 2015. 1 Haziran 2022 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ a b c "Clementia Pharmaceuticals Expands Ongoing Phase 2 Study to Include Children with Fibrodysplasia Ossificans Progressiva (FOP)" (Basın açıklaması). Clementia Pharmaceuticals. 25 Ağustos 2015. 28 Ağustos 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ a b "Pipeline". www.clementiapharma.com. 29 Eylül 2015 tarihinde kaynağından arşivlendi. Erişim tarihi: 22 Kasım 2015.
- ^ "Regeneron Shares Updates on its Ongoing FOP Research Program". International Fibrodysplasia Ossificans Progressiva Association. 9 Mart 2017. 20 Ocak 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ Lowery, J. W.; Rosen, V. (1 Temmuz 2012). "Silencing the FOP gene". Gene Therapy (İngilizce). 19 (7): 701-702. doi:10.1038/gt.2011.190. ISSN 1476-5462. 23 Ocak 2023 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ Lees-Shepard, John B.; Yamamoto, Masakazu; Biswas, Arpita A.; Stoessel, Sean J.; Nicholas, Sarah-Anne E.; Cogswell, Cathy A.; Devarakonda, Parvathi M.; Schneider, Michael J.; Cummins, Samantha M.; Legendre, Nicholas P.; Yamamoto, Shoko (2 Şubat 2018). "Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva". Nature Communications. 9: 471. doi:10.1038/s41467-018-02872-2. ISSN 2041-1723. PMC 5797136 $2. PMID 29396429. 23 Ocak 2023 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.
- ^ Williams, Eleanor; Bagarova, Jana; Kerr, Georgina; Xia, Dong-Dong; Place, Elsie S.; Dey, Devaveena; Shen, Yue; Bocobo, Geoffrey A.; Mohedas, Agustin H.; Huang, Xiuli; Sanderson, Philip E. "Saracatinib is an efficacious clinical candidate for fibrodysplasia ossificans progressiva". JCI Insight. 6 (8): e95042. doi:10.1172/jci.insight.95042. ISSN 2379-3708. PMC 8119212 $2. PMID 33705358. 3 Mayıs 2023 tarihinde kaynağından arşivlendi. Erişim tarihi: 2 Ekim 2023.