Anodonti sendromu, sürekli dişlerin oluşmadığıbir sendromdur. Kalıtsaldır, otosomal resesif yolla geçer. Süt dişleri etkilenmez.
Auriculocondylar sendrom, kalıtsal bir sendromdur. 3 fenotipi vardır. Fenotip 1 otosomal dominant (AD), fenotip 3 otosomal resesif (AR), fenotip 2 ise her iki yolla aktarılır. 1. ve 2. Fenotiplerin bulguları ortaktır; etkilenen gen farklıdır.

Plagiosefali (plagiocephaly), kafatası asimetrisiyle karakterize bir tablodur. Sagital kraniyosinostoz nedeniyle ortaya çıkan “sinostotik tip plagiosefali” olguları “gerçek plagiosefali” grubu olarak nitelendirilir; bu tür olgularda, orbitalar da asimetriktir.
Kraniyosinostoz, kraniyosinostozis (craniosynostosis), kraniyofasiyal malformasyonların ve maksillofasiyal sendromların önemli bir bölümünde etkileri görülebilen konjenital bir patolojidir. Bu olgudaki temel bulgu kafatası eklemlerinin erken kapanmasıdır; etkilediği anatomik bölgelerde ortaya çıkan malformasyonlar, hangi suturaların ne düzeyde kapanmış olmasıyla orantılıdır. Malformasyonlar genellikle etkilenen eklemin dikey yönünde belirgindir.

Baraitser-Winter sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; 2 fenotipi vardır. Genel gelişme geriliği izlenir. Kraniyosinostoz nedenli trigonosefali ve mikrosefali saptanır. Boyun kısadır. Hipertelorizm saptanır, kaş çıkıntıları yüksektedir. Göz kapakları iridir ve bilateral ptozis saptanır. Mikroftalmi ile iris ve retina defektleri (koloboma) izlenir. Kulak kepçeleri küçüktür ve aşağıdadır; işitme sorunları vardır. Burun sırtı kalın, ucu büyüktür. Uzun bir filtrum altında ince bir üst dudak bulunur. Alt dudak kalındır. Üst dudak ve damak yarıktır. Altçene geridedir (retrognati). Omurga sisteminde kifoz ve skolyoz türü malformasyonlar saptanır. Başparmak sayısı birden fazladır (polidaktili). Değişik oranlarda konjenital kalp anomalileri ve ürogenital anomaliler görülür. Beyinde corpus callosum anomalileri ile frontal lob anomalisi saptanır; epilepsi ve zeka geriliği bulguları vardır. Genel hipotoninin yanı sıra omuz kaslarında giderek artan güçsüzlük görülür. Deri lenfoması ve lösemi riski yüksektir.
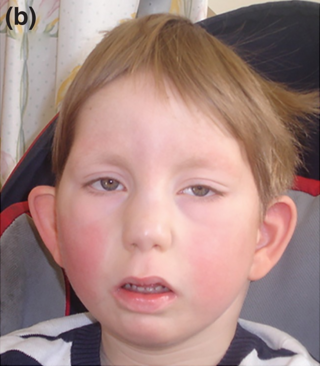
Au-Kline sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; böbrek, kalp ve yüz bulgularıyla öne çıkar.
Mikroftalmi sendromları, 18 fenotipi olan, etkilenen gen türüne göre farklı yollarla -otosomal dominant (AD), otosomal resesif (AR), X-kromozomu dominant (XLD), X-kromozomu resesif (XLR)- aktarılan kalıtsal patolojilerdir. Ortak bulgular yanı sıra farklı sistemlere özgü bulgularla da karşılaşılmaktadır. Ortak bulguların en büyük kümesi gözlerle ilgilidir.

Sotos sendromu, 3 fenotipi olan aşırı büyüme sendromlarından biridir. Sotos 1, serebral gigantizm olarak da bilinen, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Sotos 2, Malan sendromu olarak da bilinir; olguların çoğu nedensiz (spontan) olarak ortaya çıkar, kalıtsal olan birkaç olgu vardır. Sotos 3, otosomal resesif yolla aktarılan, çok ender görülen kalıtsal bir fenotiptir.

Miller-Dieker lissensefali sendromu, büyük bölümü otosomal dominant yolla aktarılan kalıtsal mikrosefali türü.

van den Ende-Gupta sendromu, otosomal resesif yolla aktarılan, yüz bulgularının ön planda olduğu kalıtsal bir sendromdur. Kraniyosinostoz nedeniyle kaniyal boşluğun ön bölümü küçüktür. Kaşlar aşağı doğrudur, göz kapak yapışıklıkları vardır. Kulaklar iridir. Burun komponentlerindeki gelişmeler yetersizdir. Yüz asimetriktir. Yanaklarda hipoplazi vardır, alt dudak kalındır. Üstçene gelişmemiştir ; aşırı çukur ve yarık bir damak saptanır. Diş dizileri bozuktur. Nazofarinks, larinks ve epiglot anomalileri saptanır.

3MC sendromu, otosomal resesif yolla aktarılan ya da spontan gen mutasyonu sonucu izole olgu olarak ortaya çıkan bir sendromdur. 3 fenotipi vardır. Önceleri Malpuech, Carnevale, Mingarelli ve Michels sendromları olarak bilinen sendromlar bu grup 3MC sendromu kapsamına alınmıştır. Göz kapakları malformasyonları, hipertelorizm, belirgin kaş çıkıntıları, kraniyofasiyal yarıklar, işitme sorunları ve algılamada güçlükler 3MC sendromunun fenotiplerinde çoğunlukla saptanan ortak bulgulardır.

Bohring-Opitz sendromu, otosomal dominant yolla aktarılan, gelişme ve zeka geriliği bulgularının ön planda olduğu, hastaların çoğunun çocukluk yaşlarında kaybedildiği bir sendromdur. C sendromu'nun fenotipi olarak benimsenir.

Cumming sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur.

Diş eksikliği sendromu, dişlerin bir bölümünün oluşmadığı, 10 fenotipi olan bir sendromdur. Diş eksiklikleri (hipodonti), çok sayıdaki sendromda bulgulardan biri olarak saptanabilir. Bu sendromdaki neredeyse tek bulgu "diş eksikliği"dir.
Roberts sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Kol ve bacak kemiklerinin eksikliği nedeniyle, eller ve ayaklar gövdeye doğrudan bağlı gibidir.
Saldino-Noonan sendromu, otosomal resesif yolla aktarılan asfiksiyan torasik displazi sendromu’nun 22 fenotipinin 3.sü olan kalıtsal bir sendromdur. Asfiksiyan torasik displazi sendromu’nun ortak bulgularına ek olarak, dilde hamartomalar, klavikulaların bisiklet gidonu biçiminde olması, cinsel organlarda malformasyonlar, enkondral ossifikasyon bozuklukları, tibia yokluğu ve skolyoz vardır.
Majewski sendromu, otosomal resesif yolla aktarılan asfiksiyan torasik displazi sendromu’nun 22 fenotipinin 6.sı olan kalıtsal bir sendromdur. Asfiksiyan torasik displazi sendromu'ndaki ortak bulguların büyük bölümü Majewski sendromu'nda da saptanmaktadır. Dudak yarığının orta çizgide olması, epiglot ve larinks malformasyonları ile klavikula ve skapula anomalileri farklı olan bulgulardır.
Mainzer-Saldino sendromu, otosomal resesif yolla aktarılan asfiksiyan torasik displazi sendromu'nun 22 fenotipinin 9.su olan kalıtsal bir sendromdur. Asfiksiyan torasik displazi sendromu'ndaki ortak bulguların büyük bölümü Mainzer-Saldino sendromu'nda da saptanmaktadır. Üçgenimsi kafatası yapısı (trigonosefali), ön-arka düzlemde uzun kafatası (skafosefali), görme sorunlarına neden olan retinitis pigmentosa, ağız açıklığının büyük olması (makrostomi), damak ve dudak yarıklarının bulunmaması, dişlerin yerleştiği kemikte düzensizlikler görece sık rastlanan farklılıklardır.
Saethre-Chotzen sendromu (acrocephalosyndactylia III), fiziksel gelişme geriliği, kraniyosinostoz nedenli kafatası anomalileri, asimetrik yüz, göz ve parmak malformasyonlarının saptandığı otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Kraniyosinostozun çok sayıda eklemi etkilediği olgularda “kafaiçi basıncı artışı sendromu (KİBAS)” gelişebilir.
Shprintzen-Goldberg sendromu , marfanoid yapı, kraniyosinostoz, kardiyovasküler ve nörolojik anomalilerle karakterize, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Marfanoid yapı, Marfan sendromundaki bulguların büyük bölümünün saptandığı, ancak Marfan sendromunudaki genetik altyapının bulunmadığı olgular için kullanılan nitelemedir; uzun kollar ve bacaklar, örümceksi parmaklar (araknodaktili), eklemlerde aşırı gevşeklik ve mitral kapak prolapsusu Marfan sendromunu andıran başlıca bulgulardır. Robbins Shprintzen-Goldberg sendromu’nun, Marfan sendromu ve Loeys-Dietz sendromu ile oldukça çok büyük benzerlikleri vardır.