
Makula dejenerasyonu ya da sarı nokta hastalığı kelime anlamı itibarı ile, makulada meydana gelen herhangi dejeneratif bir süreci tanımlasa da, bu makalede yaşa bağlı makula dejenerasyonu (YBMD) anlatılmaktadır.

Nöroloji ya da sinir bilimi, genel olarak beyin, beyin sapı, omurilik ve çevresel sinir sistemiyle kasların hastalıklarını inceleyen, cerrahi dışındaki tedavi uygulamalarını içeren tıp bilimi dalıdır. Nöroloji zamanla içine kapalı ve sınırlı bir dal olmaktan çıkmış, epilepsi, hareket bozuklukları, beyin damar hastalıkları, bunamalar, uyku bozuklukları gibi ayrıca özelleşmişlik gerektiren alt disiplinlere bölünmüştür, bunun yanı sıra 19. yüzyılda ruh hastalıklarıyla birlikte ele alınırken, 20. yüzyıldan itibaren psikiyatri ayrı bir dal olarak ayrılmıştır. Tüm bu alanlardaki ciddi laboratuvar arka planının yanı sıra günümüze nöroloji pek çok başka tıp alanı ile multidisipliner bir ilişki içindedir.
Ailevi Akdeniz ateşi veya eskiden yabancı literatürde Periyodik hastalık veya Ermeni hastalığı, sıklıkla Ermeni, Yahudi, Türk, Orta Doğu Arap toplumlarında ve Japonlarda görülen irsi ateşli hastalıktır. Ailevi Akdeniz Ateşi, tekrarlayan ateş, karın ağrısı, göğüs ağrısı ve eklem ağrısı nöbetleri yapan bir hastalıktır. Nöbetler genellikle 24-48 saat sürer. Hastalarda nöbetler dışında hiçbir belirti yoktur.
Mutasyon ya da değişinim, bir canlının genomu içindeki DNA ya da RNA diziliminde meydana gelen kalıcı değişmelerdir. Mutasyona sahip bir organizma ise mutant olarak adlandırılır.

Kromozom 18, toplamda 22 çift olan otozomal insan kromozomlarından onsekizincisidir. İnsanlarda normalde bir çift halinde bulunur. 76 milyon baz çiftine ve toplam hücre DNA'sının %2,5'ine sahiptir. Kromozom 18, muhtemelen 300 ile 400 arasında gen içermektedir.
Marfan sendromu, anormal bağ dokusu yapısı ile karakterize bir sendromdur. Birçok sistemi aynı anda etkilemektedir. Birçok hastada uzun parmaklar karakterize olmakla birlikte tesadüfen bir ağrı sonucu hastaneye giden bireylerde de teşhis konmasından da anlaşılacağı üzere fenotip üzerindeki etkisi değişkendir.

Retinitis pigmentosa (RP), halk arasında tavuk karası ve gece körlüğü adlarıyla bilinen ve görme kaybına neden olan genetik bir göz hastalığıdır. Her 4.000 kişide 1'i etkilediği tahmin edilmektedir.
Huntington hastalığı (HH), genetik, nörodejeneratif bir beyin hastalığıdır. Hastalarda bazı hareket bozukluklarının yanı sıra mental gerilik görülür. Adını 1872 yılında hastalığın kalıtsal olduğunu ilk olarak gözlemleyen Dr. George Huntington'dan alır. Otozomal dominant olarak kalıtılan bir hastalık olup beyin ve sinir sistemini etkiler. Hasta kişiler genellikle heterozigotlardır. Hastalığın ilk belirtileri 30-50 yaş arasında gözlenir. Hasta bu dönem zarfında çocuk yapmış ise çocuklarına bu hastalığı aktarma riski %50 oranındadır. Hastalık ölümcül olmasa da bu durum hastalığın gelişimine bağlıdır. Hastalığın nedeni Huntington proteinin üretim bozukluğundan dolayı ortaya çıkmaktadır.
Genetik bozukluk, genlerde ve kromozomlarda görülen anomaliler sonucu ortaya çıkan durumdur. Kanser gibi bazı hastalıklar yaşam sırasında edinilen ve bazı hücrelerde görülen genetik anomaliler nedeniyle olsa da "genetik hastalık" terimi genellikle vücuttaki tüm hücrelerde bulunan ve döllenmeden beri var olan hastalıklar için kullanılır. Bazı genetik bozukluklar, sperm ve yumurtalar gibi üreme hücrelerini oluşturan mayoz bölünme sırasında oluşan kromozom anomalileri nedeniyle ortaya çıkar. Bunlara örnek olarak Down sendromu, Turner sendromu (45X0) ve Klinefelter sendromu sayılabilir. Diğer genetik değişiklikler ebeveynler tarafından tohum hücrelerin oluşturulması sırasında ortaya çıkabilir. Bunlara bir örnek frajil X sendromu ya da Huntington hastalığına neden olabilen üçlü yayılma tekrar mutasyonudur. Hatalı genler ebeveynlerden olduğu gibi alınmış da olabilir. Bu genellikle sağlıklı ama resesif gen taşıyan iki kişinin üremesi ya da hatalı genin dominant olması sonucunda olabilir.
Kistik fibrozis, akciğer, pankreas, bağırsak, ter bezleri dış salgı bezlerinde görülen, otozomal resesif kalıtımlı bir gen hastalığıdır. Kistik fibrozis hastalığı, aynı anda solunum sistemi, sindirim sistemi gibi vücudun birden çok sistem ve organını etkileyebilir. Doğumla birlikte görülen fibrozis, bu etkileme sonucu işlev bozukluklarına neden olur.
Metakromatik lökodistrofi, Aril Sülfataz A enzim eksikliğnden kaynaklanan bir lizozomal depo hastalığıdır.
Hunter sendromu ya da Tip II mukopolisakkaridoz, iduronat-2-sülfataz (I2S) enziminin eksikliğinden ya da yokluğundan kaynaklanan lizozomal depo hastalığıdır. Doktor Charles A. Hunter (1873-1955) tarafından ilk kez 1917 yılında tanımlandığından, Hunter sendromu olarak adlandırılır.
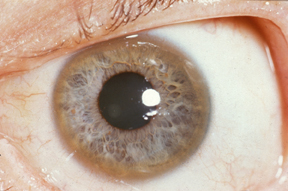
Wilson hastalığı veya hepatolentiküler dejenerasyon dokularda bakır birikimine yol açan otozomal resesif geçişli genetik bir hastalıktır. Bu hastalık kendini psikiyatrik veya nörolojik belirtilerle ve karaciğer hastalığıyla gösterir. Hastalığı ilaçla tedavi etmek mümkündür.

Orak hücreli anemi, alyuvarlardaki oksijen taşıyıcı protein olan hemoglobinin anormalliği sonucu alyuvarların orak şeklini almasıyla oluşan otozomal resesif kalıtılan genetik bir hastalıktır.
Donohue sendromu, otosomal resesif yolla aktarılan, nadir bir kalıtsal sendromdur. Leprechaunizm ismi, hastalıktan muzdarip olanların çoğu kez elfvari özelliklere sahip olmaları ve olağandan küçük olmalarından gelir (cüce cin sendromu). Ön planda, insülin reseptör defektine bağlı insülin resistansı bulguları saptanır. Pankreasta adacık beta hücrelerinin hiperplazisi nedeniyle kan insülin düzeyi yüksektir (hiperinsülinemi). Spontan abortus ve bebek ölümü riski oldukça yüksektir.
Otozomal dominant geçer, kalıtsaldır, otozomal resesif formları da vardır. Kas membranı klor kanallarında bozukluk vardır. 10-20 yaşlarında başlar ve tek semptom myotonidir. Myotoni, kasların kasıldıktan sonra hızlıca gevşememesi olarak tanımlanır. Myotoniye iyon kanallarının çalışmasından sorumlu olana gende meydana gelen bir mutasyon sebep olmaktadır. Myotoni genelde tüm kasları tutar fakat göz kapağında ve ellerde belirgindir. Myotoni soğukta ve hareketsizlikle artar, egzersizle azalır. Kaslarında yaygın hipertrofi vardır ve bu hastaya Herkül görünümü verir. Yaş ilerledikçe semptomlarda hafifleme olabilir. Benign seyirlidir. Resesif form, dominanta göre iki kez daha fazla görülür ve daha ağır seyreder. Distal kaslarda ılımlı atrofi ve kuvvetsizlik ortaya çıkabilir.

Genetik hastalıklar , bir ailede kuşaktan kuşağa aktarılabilen patolojileri niteleyen tanımlamadır. Kalıtsal hastalıkların gelecek kuşaklara aktarılmasında etkili olan faktörlerler, genlerdeki ve kromozomlardaki yapısal değişikliklerdir.
Marinesco–Sjögren sendromu (MSS), bazen Marinescu-Sjögren sendromu olarak da adlandırılır, nadir görülen otozomal resesif bir hastalıktır.

OTC eksikliği olarak da bilinen ornitin transkarbamilaz eksikliği, insanlarda en yaygın görülen üre döngüsü bozukluğudur. Ornitin transkarbamilaz, bu bozukluktaki kusurlu enzim, üre döngüsünün proksimal kısmındaki son enzimdir ve karbamoil fosfat ve ornitini sitrüline dönüştürmekten sorumludur. OTC eksikliği, X'e bağlı otozomal resesif bir şekilde kalıtılır, yani erkekler kadınlardan daha sık etkilenir.
Albinizm-siyah saç teli-hücre göç bozukluğu, bir bireyin fiziksel görünümünü ve fizyolojisini etkileyen durumları tanımlayan terim ve kavramların kısaltmasıdır: (1) A – albinizm, (2) B – siyah saç teli, (3) C – bağırsak nörositlerinin hücre göç bozukluğu ve (4) D – sinirsel tip işitme kaybı. Bu sendrom, endotelin B reseptör geni (EDNRB) mutasyonundan kaynaklanır.