
Beyin bazal gangliyonların hastalığıdır.
Opitz GBBB sendromu 22q11.2 sendromları kümesinin kalıtsal olan tek fenotipidir. 2 tip Opitz GBBB sendromu bilinmektedir.
Ağız-Yüz-Parmak sendromu , günümüze dek 16 fenotipi belirlenmiş olan bir sendromlar kümesidir. Bilinen fenotiplere yenileri eklenebilir. Ağız-Yüz-Parmak sendromunun OFD arasında en sık ratlanılanı OFP tip I temel bulguları içerir; sıkça rastlanan öteki fenotiplerde, tip I'e eklenen ya da çıkarılan yan bulgular vardır.
Holoprosensefali, fetüste, beyin ön bölgesi (prosencephalon) gelişiminin aksaması ve loblarına ayrılamaması olgusudur; ek olarak, mikrosefali, hipofiz bezi ön lobunun agenezi ya da displazisi, yüzde orta çizgi anomalileri, burun malformasyonları, yarık dudak-yarık damak saptanır. 4 tipi vardır:
- Lober tip: Beyin ön lobları oluşmamıştır. En güçlü tiptir.
- Semilober tip: Beyin ön loblarının bir bölümü oluşmuştur, iki hemisfer arasında silik bir çöküntü bulunur.
- Sintelensefali (Syntelencephaly) tipi: Beyin ön lobları hipoplaziktir; ancak parietal loblarla arasında bulunması gereken oluk seçilemez.
- Corpus callosum agenezi içeren tip: Corpus callosum hiç yoktur ya da hipoplaziktir.

Agnati-Otosefali kompleksi, öncelikle mikrognatiye neden olan malformasyonlardan biridir; altçene yokluğu anlamına gelse de, altçenenin tümü değil ancak bir bölümünün eksikliği ya da oldukça aşırı bir hipoplazisi söz konudur. Ender görülen bu kompleksin kalıtsal olabileceği gibi, gen mutasyonlarının ve teratojenlerin etkisini gösteren olgular da bildirilmiştir.
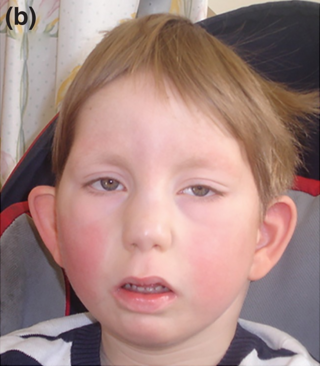
Au-Kline sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; böbrek, kalp ve yüz bulgularıyla öne çıkar.
Frontonazal displazi, otosomal resesif yolla aktarılan, alın ve burun bölgelerinin malformasyonlarıyla karakterize kalıtsal bir sendromdur; ALX3 genindeki mutasyonun sonucudur. En önemli bulgu, frontal kemikte ve yüz kemiklerinde orta çizgi yarıklarının bulunmasıdır ile bunun sonucunda ortaya çıkan nazofrontal ensefalosel’dir. Alın derisinde lipoma olabilir. Mikroftalmi saptanır, gözler birbirinden aşırı uzaktadır (hipertelorizm); ptozis, kapak bileşkelerinde anomaliler, katarakt ve epibulbar dermoidler belirlenir. Kulak kepçeleri aşağıdadır, işitme sorunları vardır. Burun kökü aşırı yayvandır; bu alanda saptanan malformasyonların en önemlisi yarıktır. Üstçenede, paranazal sinüslerde ve frontal sinüslerde hipoplazi izlenir. Kalpte Fallot tetralojisi saptanabilir. Pektoral kaslar hipoplazi ya da agenez olabilir. El parmakları kısa ve kıvrıktır. Beyinde Corpus callosum anomalileri saptanır, zeka geriliği vardır. Akromelik frontonazal disostoz fenotipindeki bulgular daha azdır.
Mikroftalmi sendromları, 18 fenotipi olan, etkilenen gen türüne göre farklı yollarla -otosomal dominant (AD), otosomal resesif (AR), X-kromozomu dominant (XLD), X-kromozomu resesif (XLR)- aktarılan kalıtsal patolojilerdir. Ortak bulgular yanı sıra farklı sistemlere özgü bulgularla da karşılaşılmaktadır. Ortak bulguların en büyük kümesi gözlerle ilgilidir.

Fryns sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Fetüsün amnion sıvısı fazladır (polihidramnios) ve bebek iridir (makrosomi); yenidoğan ölümleri sıktır.

Schinzel-Giedion sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Güçlü bir zeka geriliği ile birlikte garip bir yüz yapısı ve çok sayıda doğumsal anomaliler görülür. Cüceliğe yakın düzeyde boy kısalığına neden olan genel gelişme geriliği vardır.

MVA sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. 3 fenotipi vardır; fenotip 1 en sık görülenidir. Anöploidi olarak niterlendirilen kromozom sayısı anomalisinden kökenlidir; kromozom eksikliği (monosomi) ya da fazlalığı (trisomi) vardır. Ancak, bazı hücrelerdeki kromozom sayısının normal olması nedeniyle “mozaisizm” grubu içinde yer alır.

Schinzel acrocallosal sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Joubert sendromunun önemli fenotiplerindendir. Santral sinir sisteminde corpus callosum yetersizliği ve zeka geriliği bulguları ile parmaklarda belirgin olan çok sayıda oluşum kusurları saptanır.

Serebrokostomandibular sendrom , otosomal dominant yolla aktarılan kalıtsal bir sendromdur; çok az sayıda otosomal resesif olgular da vardır. Bebeklerin yarıya yakın bölümü 1 yaşından önce kaybedilir. Doğum ölçümleri normaldir, ancak zamanla gelişme geriliği bulguları belirir.
Toriello-Carey sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Doğum ölçümleri normaldir, ancak zamanla gelişme geriliği bulguları belirir.

Hartsfield sendromu, otosomal dominant yolla aktarılan, ender görülen, kalıtsal bir sendromdur. El ve ayak parmaklarının konjenital eksikliği (ektrodaktili), yarık dudak ve yarık damak ile kafatası malformasyonu (holoprosensefali) üçlüsü temel bulgulardır.

Pai sendromu, otosomal dominant yolla aktarılan, ender görülen, kalıtsal bir sendromdur. Yüz derisinde papillomatöz oluşumlar, burun polipleri, üst dudakta orta çizgi yarığı, göz anomalileri ile beyinde orta çizgide ve corpus callosum çevresinde yağ dokusu uru bulguları ön plandadır.

Klippel-Feil sendromu, otosomal dominant yolla aktarılan, kalıtsal bir sendromdur. 4 fenotipi vardır; fenotip 2,3 ve 4 çok enderdir. Temel bulgu, Klippel-Feil anomalisidir; bu tabloda, servikal vertebralarda saptanan hipoplazi ve kaynaşma görülür. Servikal vertebralardaki anomali nedeniyle boyun kısa ve hareketleri sınırlıdır (torticollis). Trapez kasında yelkensi görünüm izlenir. Servikal spinal kanalda darlık, spina bifida ve skolyoz görülebilir. Skapula yukarı doğru yer değiştirmiştir. Ensedeki düşük saç çizgisi oldukça aşağıda gibi algılanır.

Peters’ Plus sendromu , otosomal resesif yolla aktarılan, kalıtsal bir sendromdur. Genel gelişme geriliği nedeniyle boy kısadır. Boyun kalın, alın bombesi yüksektir. Yüz derisinde aşırı kıllanma (hipertrikoz) olabilir. Gözler birbirilerinden uzakçadır (hipertelorizm), göz kapaklarının açıklığı dardır. Görme sorunlarına neden olan Peters anomalisi, glokom, katarakt ve retinafs defektler (koloboma) vardır. İşitme sorunları saptanır; kulaklar küçük ve geridedir, kulak deliği önündeki deride çukurlar görülür. Üst dudak kabarıktır. Yarık dudak ve yarık damak saptanır. Altçene küçüktür (mikrognati). Üst yan kesici dişler eksiktir (hipodonti). Dili ağız tabanına bağlayan bağ kısadır (ankyloglossia).

Tetra-amelia sendromu, kol ve bacakların oluşamadığı, skapula ve klavikula malformasyonlarının saptandığı, otosomal resesif olarak (XLR) aktarılan bir sendromdur. Doğumdan sonraki ilk haftalarda ölüm riski çok yüksektir.

Vici sendromu, psikomotor gelişme geriliğinin yanı sıra göz, dolaşım sistemi ve bağışıklık sistemindeki defektlerle karakterize, otosomal resesif yolla aktarılan kalıtsal bir sendromdur.
