
Coffin-Siris sendromu, ektodermal displazi bulgularını da içeren, otosomal dominant geçen kalıtsal bir sendromdur. 8 fenotipi vardır. Çene-yüz bulguları fenotiplerden 3'ünde belirgindir. Hastalarda genel gelişme geriliği vardır. Kafatası küçüktür (mikrosefali).
Gorlin-Cohen sendromu , ektodermal displazi bulgularını da içerebilen bir OPD sendromu tipidir. 2 fenotipi vardır:
- Frontometaphyseal dysplasia 1
- Frontometaphyseal dysplasia 2
Treacher Collins sendromu, yüz bulgularının ön planda olduğu kalıtsal bir sendromlardan biridir. Otosomal dominant yolla aktarılan 3 fenotipi vardır; bunlardan fenotip 2’nin otosomal resesif yolla da aktarılabildiği bilinmektedir. Fenotip 2, Treacher Collins-Franceschetti sendromu olarak da bilinir. Mandibulofacial dysostosis sendromu tiplerinden biri olarak niteleyen uzman görüşü vardır.

Baraitser-Winter sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; 2 fenotipi vardır. Genel gelişme geriliği izlenir. Kraniyosinostoz nedenli trigonosefali ve mikrosefali saptanır. Boyun kısadır. Hipertelorizm saptanır, kaş çıkıntıları yüksektedir. Göz kapakları iridir ve bilateral ptozis saptanır. Mikroftalmi ile iris ve retina defektleri (koloboma) izlenir. Kulak kepçeleri küçüktür ve aşağıdadır; işitme sorunları vardır. Burun sırtı kalın, ucu büyüktür. Uzun bir filtrum altında ince bir üst dudak bulunur. Alt dudak kalındır. Üst dudak ve damak yarıktır. Altçene geridedir (retrognati). Omurga sisteminde kifoz ve skolyoz türü malformasyonlar saptanır. Başparmak sayısı birden fazladır (polidaktili). Değişik oranlarda konjenital kalp anomalileri ve ürogenital anomaliler görülür. Beyinde corpus callosum anomalileri ile frontal lob anomalisi saptanır; epilepsi ve zeka geriliği bulguları vardır. Genel hipotoninin yanı sıra omuz kaslarında giderek artan güçsüzlük görülür. Deri lenfoması ve lösemi riski yüksektir.
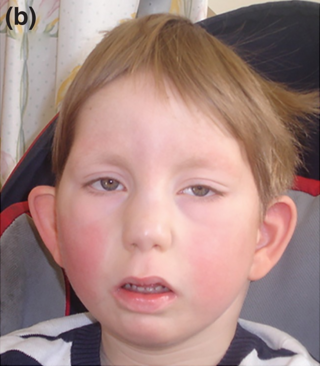
Au-Kline sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; böbrek, kalp ve yüz bulgularıyla öne çıkar.

Cornelia de Lange sendromu , 5 fenotipi olan kalıtsal bir sendromdur. Tüm fenotiplerde benzer bulgular saptanır; farklılık etkilenen genlerdedir.
Frontonazal displazi, otosomal resesif yolla aktarılan, alın ve burun bölgelerinin malformasyonlarıyla karakterize kalıtsal bir sendromdur; ALX3 genindeki mutasyonun sonucudur. En önemli bulgu, frontal kemikte ve yüz kemiklerinde orta çizgi yarıklarının bulunmasıdır ile bunun sonucunda ortaya çıkan nazofrontal ensefalosel’dir. Alın derisinde lipoma olabilir. Mikroftalmi saptanır, gözler birbirinden aşırı uzaktadır (hipertelorizm); ptozis, kapak bileşkelerinde anomaliler, katarakt ve epibulbar dermoidler belirlenir. Kulak kepçeleri aşağıdadır, işitme sorunları vardır. Burun kökü aşırı yayvandır; bu alanda saptanan malformasyonların en önemlisi yarıktır. Üstçenede, paranazal sinüslerde ve frontal sinüslerde hipoplazi izlenir. Kalpte Fallot tetralojisi saptanabilir. Pektoral kaslar hipoplazi ya da agenez olabilir. El parmakları kısa ve kıvrıktır. Beyinde Corpus callosum anomalileri saptanır, zeka geriliği vardır. Akromelik frontonazal disostoz fenotipindeki bulgular daha azdır.

Joubert sendromu, kalıtsal bir sendromdur; 36 fenotipinden 34'ü otosomal resesif yolla aktarılır, olguların bir bölümü Meckel sendromu ile çakışmaktadır. Joubert sendromunun tüm fenotiplerinde 3 temel bulgu vardır: santral sinir sistemi anomalileri, fizik gelişme geriliği, solunum sorunları. Serebellum (beyincik) vermisi ve beyin sapı hipoplazisi ile beyin radyolojisinde saptanan “azıdişi bulgusu ” santral sinir sistemi anomalilerinin başında gelir. Ataksi ve hipotoni sık görülür. Bazı hastalarda oksipital meningosel ya da meningomyelosel olabilir. Schinzel acrocallosal sendromu, önemli fenotiplerinden biridir.

Kabuki sendromu, iki fenotipi olan bir sendromdur: Kabuki sendromu 1 ve Kabuki sendromu 2 (Kabuk2). Kabuk1, otosomal dominant yolla aktarılan kalıtsal ya da izole olgular biçiminde görülür; Kabuk2 ise, X-kromozomu aracılığıyla dominant yolla (XLD) aktarılır. Her iki fenotipini çok sayıda ortak bulguları vardır; özellikle yüz, iskelet sistemi, gelişme geriliği, parmakizleriyle ilgili bulgular ve zeka geriliği önemlidir.

Sotos sendromu, 3 fenotipi olan aşırı büyüme sendromlarından biridir. Sotos 1, serebral gigantizm olarak da bilinen, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Sotos 2, Malan sendromu olarak da bilinir; olguların çoğu nedensiz (spontan) olarak ortaya çıkar, kalıtsal olan birkaç olgu vardır. Sotos 3, otosomal resesif yolla aktarılan, çok ender görülen kalıtsal bir fenotiptir.

Robinow sendromu, 5 fenotipi olan kalıtsal bir sendromdur; fenotiplerin ikisi otosomal resessif, üçü otosomal dominant yolla aktarılır. Tüm fenotiplerde iskelet sistemi bulguları yoğun olmakla birlikte maksillofasiyal bulguları ile her iki cinste de saptanan genital organ hipoplazileri ortak bulgulardır.

Multipl pterygium sendromu (Escobar sendromu), otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Lethal türünde fetüsün yaşama yetisi yoktur.
van den Ende-Gupta sendromu, otosomal resesif yolla aktarılan, yüz bulgularının ön planda olduğu kalıtsal bir sendromdur. Kraniyosinostoz nedeniyle kaniyal boşluğun ön bölümü küçüktür. Kaşlar aşağı doğrudur, göz kapak yapışıklıkları vardır. Kulaklar iridir. Burun komponentlerindeki gelişmeler yetersizdir. Yüz asimetriktir. Yanaklarda hipoplazi vardır, alt dudak kalındır. Üstçene gelişmemiştir ; aşırı çukur ve yarık bir damak saptanır. Diş dizileri bozuktur. Nazofarinks, larinks ve epiglot anomalileri saptanır.

Bohring-Opitz sendromu, otosomal dominant yolla aktarılan, gelişme ve zeka geriliği bulgularının ön planda olduğu, hastaların çoğunun çocukluk yaşlarında kaybedildiği bir sendromdur. C sendromu'nun fenotipi olarak benimsenir.

Meckel-Gruber sendromu, çok sayıda genin etkilenmesinden kaynaklanan, bu nedenle 15 fenotipi olan, otosomal resesif yolla aktarılan, siliyopatiler grubundan, kalıtsal bir sendromdur. Kistik böbrekler, beyin anomalileri ve ensefalosel, polidaktili ve karaciğer anomalileri başlıca bulgulardır. En sık görülen fenotip Meckel sendromu 1, öteki fenotiplerdeki bulguların çok büyük bir bölümünü içerir. Bazı fenotipleri Joubert sendromuyla çakışan nitelikleri içermektedir.
Shprintzen-Goldberg sendromu , marfanoid yapı, kraniyosinostoz, kardiyovasküler ve nörolojik anomalilerle karakterize, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Marfanoid yapı, Marfan sendromundaki bulguların büyük bölümünün saptandığı, ancak Marfan sendromunudaki genetik altyapının bulunmadığı olgular için kullanılan nitelemedir; uzun kollar ve bacaklar, örümceksi parmaklar (araknodaktili), eklemlerde aşırı gevşeklik ve mitral kapak prolapsusu Marfan sendromunu andıran başlıca bulgulardır. Robbins Shprintzen-Goldberg sendromu’nun, Marfan sendromu ve Loeys-Dietz sendromu ile oldukça çok büyük benzerlikleri vardır.

Snyder-Robinson sendromu, “mental retardasyon sendromu”nun 54 fenotipinden biri olan, X-kromozomu aracılığıyla resesif (XLR) yolla aktarılan kalıtsal bir sendromdur. Ayrıca, marfanoid yapı, kas ve iskelet sistemi anomalileri ve çene-yüz bulguları vardır. Marfanoid yapı, Marfan sendromundaki bulguların büyük bölümünün saptandığı, ancak Marfan sendromunudaki genetik altyapının bulunmadığı olgular için kullanılan nitelemedir; uzun kollar ve bacaklar, örümceksi parmaklar (araknodaktili), eklemlerde aşırı gevşeklik ve mitral kapak prolapsusu Marfan sendromunu andıran başlıca bulgulardır.
BOR sendromu, otosomal dominant aktarılan kalıtsal bir sendromdur.
HMC sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Sendroma adını veren 3 temel bulgu vardır: Hipertelorizm-küçük kulak deliği (microtia)-yüz yarıkları. Mikrosefali ve hipertelorizm belirgindir. Kulak boşlukları dardır ve işitme güçlüğü vardır. Burun sırtı yayvandır. Ağız açıklığı küçüktür (mikrostomi); yüz, burun, dudak ve damak yarıkları bulunur. Konjenital kalp defektleri, böbreklerde yer değişikliği, vertebra anomalileri saptanır. Serçe parmağı kısadır, parmaklarda yapışıklıklar vardır. Psikomotor gelişme geriliği görülür.
Meckel sendromu, birden fazla etkisi olan bir genle ilgili (pleiotropic), otosomal resesif yolla aktarılan, 15 kadar fenotipi bulunan kalıtsal bir sendromdur. Çok sayıda ve karmaşık bulguların varlığı tanıda güçlükler oluşturur. Bazı uzmanlar, Meckel sendromunun 3 temel bulgusu olduğunu savunurlar: (i) Santral sinir sistemi malformasyonları ; (ii) Kistik böbrekler; (iii) Karaciğer patolojileri. Bebeklerin yaşama yetisi çok kısıtlıdır; canlı doğabilenlerin çoğu birkaç hafta içimde yitirilir.
