Cleidocranial dysostosis, kemik malformasyonları içeren kalıtsal bir sendromdur; ebeveynlerden birinden otosomal dominant yolla gelir. Genetik incelemeler, kemik gelişimiyle ilgili RUNX2 genindeki bir sorunu göstermektedir. 3 tipi vardır; dişlerin etkilenmesi bunlardan yalnızca birinde saptanır. İskelet sistemi etkilenmesinin en tipik bulgusu köprücük kemiklerinde (klavikula;clavicula) görülür. Köprücük kemikleri ya hiç oluşmamıştır (agenezis) ya da gelişmesi yetersizdir (hipoplazi); bu nedenle, hastalar, omuzlarını orta çizgi üzerinde birleştirebilirler. Kafatasındaki bıngıldaklar ve pelvisteki simfizi uzun süre kapanamaz.
Hipohidrotik ektodermal displazi sendromu , derideki yağ ve ter bezlerinin yokluğu sonucunda ortaya çıkan ”terleme azlığı/yokluğu (hipohidroz)” ile öne çıkan bir ektodermal displazi türüdür; 16 fenotipi vardır. Hipohidroz, çocuk hastalarda vücut ısısında yükselme (hipertermi) ataklarına neden olabilir; santral sinir sistemi etkilenmesine bağlı havale tablosu gelişebilir.
Kraniyoektodermal displazi (Cranioectodermal dysplasia), ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur. 4 fenotipi vardır:
- Cranioectodermal dysplasia 1 (Sensenbrenner sendromu; Levin sendromu; arthrodentoosseous dysplasia)
- Cranioectodermal dysplasia 2
- Cranioectodermal dysplasia 3
- Cranioectodermal dysplasia 4
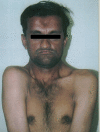
Oculodentodigital sendrom (ODDD; oculodentoosseous dysplasia), ektodermal displazi bulguları da içeren, otosomal dominant geçen kalıtsal bir sendromdur. Bazı olgular, otosomal resesif geçiş gösterebilir ya da ileri yaş gebeliklerden doğan bebeklerdeki spontan gen mutasyonunun sonucudur.
Ağız-Yüz-Parmak sendromu , günümüze dek 16 fenotipi belirlenmiş olan bir sendromlar kümesidir. Bilinen fenotiplere yenileri eklenebilir. Ağız-Yüz-Parmak sendromunun OFD arasında en sık ratlanılanı OFP tip I temel bulguları içerir; sıkça rastlanan öteki fenotiplerde, tip I'e eklenen ya da çıkarılan yan bulgular vardır.
Ağız-Yüz-Parmak sendromu tip 1 , ektodermal displazi bulguları da içeren, X-kromozomu aracılığıyla dominant (XLD) geçen kalıtsal bir sendromdur. Simpson-Golabi-Behmel sendromu tip 2 ile alelik bağı olduğu belirlenmiştir. Kız çocuklarında görece sıktır. Erkek fetüsler, kalp ve beyin anomalilerinin neden olduğu intauterin ölümler nedeniyle kaybedilirler. Belirgin bir genel gelişme geriliği saptanır.
Oral-Facial-Digital sendrom II (orofaciodigital sendrom 2, Mohr sendromu; Mohr-Claussen sendromu), ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur. Belirgin bir genel gelişme geriliği saptanır.
OPD sendromu (otopalatodigital sendrom), X-kromozomu aracılığıyla dominant (XLD) yolla geçen kalıtsal bir sendromdur (Gorlin-Cohen sendromu hariç). Kız çocukları daha hafif etkilenir. Tümü benzer bulgular içeren 5 tipi vardır:
- Otopalatodigital sendrom tip I (OPD tip1)
- Otopalatodigital sendrom tip II (OPD tip2)
- Gorlin-Cohen sendromu (ilgili sayfaya gidiniz)
- Melnick-Needles sendromu (tip 1 ile allelik bağlantısı vardır; ilgili sayfaya gidiniz)
- Terminal osseöz displazi ve pigmentli deri defektleri (çok enderdir, ayrıntı verilmemiştir).
Gorlin-Cohen sendromu , ektodermal displazi bulgularını da içerebilen bir OPD sendromu tipidir. 2 fenotipi vardır:
- Frontometaphyseal dysplasia 1
- Frontometaphyseal dysplasia 2

Marshall sendromu, ektodermal displazi bulguları da içerebilen, otosomal dominant yolla geçen kalıtsal bir sendromdur.

Bosma arini mikroftalmi sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; göz ve burun bulguları ön plandadır.

Cornelia de Lange sendromu , 5 fenotipi olan kalıtsal bir sendromdur. Tüm fenotiplerde benzer bulgular saptanır; farklılık etkilenen genlerdedir.

Robinow sendromu, 5 fenotipi olan kalıtsal bir sendromdur; fenotiplerin ikisi otosomal resessif, üçü otosomal dominant yolla aktarılır. Tüm fenotiplerde iskelet sistemi bulguları yoğun olmakla birlikte maksillofasiyal bulguları ile her iki cinste de saptanan genital organ hipoplazileri ortak bulgulardır.

Sipondilokarpotarsal sinoztozis sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. İskelet sisteminin tümünü ilgilendiren anomalilerin yanı sıra, özellikle gövde ve omurga sisteminin gelişmesindeki gerilik nedeniyle kısa boy ve orantısız bir vücut yapısı vardır. Parmak eklemlerinde kaynaşmalar görülür. Skapula, sternum ve kaburga anomalileri izlenir; çok sayıdaki vertebta anomalileri nedeniyle skolyoz ve lordoz saptanır. Toraksı olumsuz biçimde etkileyen çevre kemik anomalilerinin yol açtığı restriktif akciğer hastalığına bağlı solunum sorunları yaşarlar. Uzun kemiklerin epifizleri gelişememiştir ve eklemleri gevşektir. Dirsek eklemi hareketleri çok kısıtlıdır.

Fraser sendromu, otosomal resesif yolla aktarılan, 3 fenotipi olan, kalıtsal bir sendromdur; göz kapaklarının birbirlerine yapışık olması (kriptoftalmi), yapışık parmaklar (sindaktili) ile solunum-üriner-genital sistemlerin anomalileri başlıca bulgulardır.

Genitopatellar sendrom, otosomal dominant yolla aktarılan, kalıtsal bir sendromdur. Mikrosefali ve mikrognati vardır. Saçlar seyrektir. Çekik ve iri gözler, iri bir burun içeren kaba yüz yapısı saptanır. İşitme sorunlarının yanı sıra yarık damak ve dişlerin sürmesinde aksamaları olduğu görülür.
Roberts sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Kol ve bacak kemiklerinin eksikliği nedeniyle, eller ve ayaklar gövdeye doğrudan bağlı gibidir.

Hajdu-Cheney sendromu (arthrodentoosteodysplasia), iskelet sistemindeki defektlerle karakterize otosomal dominant yolla aktarılan kalıtsal bir sendromdur.
Pallister-Killian sendromu, kalıtsal olmayan, spontan olarak ortaya çıkan bir sendromdur. Boyun kısa ve kalın, alın bombesi yüksektir. Hipertelorizm saptanır. Saçlar, kaşlar ve kirpikler seyrek, göz kapakları düşük (ptozis) ve şiştir. Gözler fırlaktır (ekzoftalmi). Strabismus ve katarakt vardır. Retinada mozaiksi pigmentasyon saptanır. Burun sırtı basık ve kısadır. Kulaklar büyüktür, işitme sorunları vardır. Yanaklar dolgundur. Dudak bileşkeleri (kommisuralar) aşağı dönüktür. Üst dudak ince, alt dudak kalındır. Altçene küçüktür (mikrognati). Dil büyüktür (makroglossi). Çukur damak, yarık damak ve yarık küçükdil saptanır. Dişlerin sürmesinde aksamaları olabilir.

Wolf-Hirschhorn sendromu, genel gelişme geriliği, kafa ve yüz malformasyonları ve epileptiform atakların görüldüğü, “4. kromozomun kısa kolunun distal kısmında delesyon (4p-)” olarak bilinen kromozom anomalisi sonucu ortaya çıkan izole olgulardır. Kız hastalar görece sıktır; 1/3’ü iki yaşına ulaşamadan kaybedilir.