
Trombosit veya kan pulcukları, kan pıhtılarının oluşumunda görev alan hücre parçalarına verilen isimdir. Platelet olarak da adlandırılır. Düşük trombosit seviyeleri veya fonksiyon anormallikleri (disfonksiyon) kanamaya yatkınlığı artırırken, yüksek trombosit seviyeleri -çoğunlukla asemptomatik- tromboz riskini yükseltir.
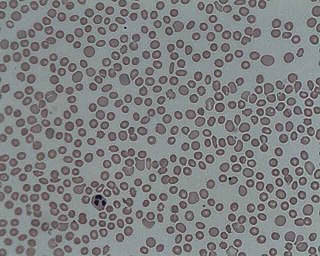
Trombositopeni veya trombopeni kandaki trombosit sayısının azlığına verilen isimdir. Bu tanımdan da anlaşılacağı gibi trombositopeni bir hastalıktan öte bir durum, bulgudur.

Korpus kallozum, beynin her iki hemisferi arasındaki bilgi iletişimini sağlayan sinir ağlarından oluşan yapıdır.

Kromozom 15, toplamda 22 çift olan otozomal insan kromozomlarından onbeşincisidir. İnsanlarda normalde bir çift halinde bulunur. 100 milyon baz çiftine ve toplam hücre DNA'sının %3 ya da %3,5'ine sahiptir. Kromozom 15, muhtemelen 700 ile 900 arasında gen içermektedir.
Yaygın gelişimsel bozukluklar (YGB), sosyalleşme ve iletişim gibi çoklu temel fonksiyonların gelişmesinde gecikmeler içeren beş bozukluğu içeren bir tanı grubudur. En çok bilinen YGB (1) otizmdir, diğer YGB’ler (2) Rett sendromu, (3) çocukluğun dezintegratif bozukluğu, (4) Asperger sendromu ve (5) başka türlü adlandırılamayan yaygın gelişimsel bozukluktur (YGB-BTA).
Çocukluğun dezintegratif bozukluğu (ÇDB) ya da Heller sendromu, dezintegratif psikoz, üç yaşından sonra çocukların dil, sosyal işlev ve motor becerilerinin gelişiminde gecikmeler olarak görülen ve ender rastlanan bir durumdur. Araştırmacılar bu durumun nelerden kaynaklandığını henüz bulamamıştır.
Mental Bozuklukların Tanısal ve Sayımsal El Kitabı veya Ruhsal Bozuklukların Tanısal ve İstatistiksel El Kitabı, zihinsel hastalıklar için bir tanı ölçütüdür. Amerikan Psikiyatri Birliği tarafından yayımlanır. İlk defa 1952'de yayımlanmıştır. Son baskısı Mart 2022'de yayımlanan DSM-5-TR'dir. 2000 yılından bu yana kullanılmakta olan bir önceki baskı DSM-IV-TR'ye göre en belirgin değişiklikler Şizofreni ve Travma Sonrası Stres Bozukluğu bölümlerinde yapılmıştır. Ayrıca DSM-5'te "Eksen Sistemi" kaldırılmıştır.
Angelman sendromu ilk olarak 1965 yılında İngiliz doktor Harry Angelman tarafından tanımlanmış nörogenetik bir bozukluktur. Irklarda görülme hızı çok iyi bilinmemekle beraber yaklaşık ensidansın 15,000 ila 30,000 canlı doğumda bir olduğu kabul edilmektedir. Anneden gelen kromozom 15'teki bir bozukluktan kaynaklandığı sanılmaktadır. Hastalığın temel bulguları zeka geriliği, yürüyüş-koordinasyon bozukluğu, konuşma bozukluğu, konvülsiyon ve uygunsuz gülümsemelerdir. Hatta bu sebeple hastalık bazen “mutlu kukla ” sendromu olarak da bilinir.

Sarhoşluk, fazla miktarda alkollü içecek tüketiminden kaynaklanan durum. Saldırganlık, mutluluk duygusu, duyularda, hareketlerde, konuşmada bozukluklar sarhoşluk durumuna eşlik edebilir.
Hipoplazi ya da az gelişim, bir organın yetersiz gelişme nedeniyle doğumsal olarak küçük kalmasıdır. Organ, tüm anatomik özelliklerini taşır, fizyolojik işlevlerini yapabilir. Körelme, irileşim ve aşırı gelişim az gelişimin (hipoplazi) karşıtı olan kavramlardır; bu olguların tümü edinseldir. Hipoplazi kavramına en yakın olan olgu aplazi olgusudur; hipoplazi ve aplazi doğumsal (konjenital) patolojilerdir.
Hunter sendromu ya da Tip II mukopolisakkaridoz, iduronat-2-sülfataz (I2S) enziminin eksikliğinden ya da yokluğundan kaynaklanan lizozomal depo hastalığıdır. Doktor Charles A. Hunter (1873-1955) tarafından ilk kez 1917 yılında tanımlandığından, Hunter sendromu olarak adlandırılır.
Bu listede önemli ve sık görülen nörolojik bozukluklar, semptomlar, bulgular ve sendromlar alfabetik olarak sıralanmıştır.
Afazi (Türkçe: söz yitimi), beynin bazı bölgelerinde meydana gelen işlev bozukluğu sonucu ortaya çıkan konuşma, konuşulanı anlama, tekrarlama, okuma-yazma gibi becerilerin gerçekleştirilememesi sorunu.
Zellweger sendromu, peroksizomlarda birtakım işlev bozukluklarının görüldüğü, hepatik ve ürogenital sistemlerin bozulması ile ilerleyen hastalığa verilen isimdir. Otozomal resesif olarak kalıtılan bu hastalığın diğer adı serebro-hepato-renal sendromdur.

XXYY sendromu, erkeklerin fazladan bir X ve Y kromozomlarının bulunduğu eşey kromozomları bozukluğu. İnsan hücreleri genellikle anne ve babadan olmak üzere iki cinsiyet kromozomu içerir. Genellikle dişiler iki X kromozomuna (XX), erkekler bir X bir Y kromozomuna (XY) sahiptir. Düzgün çalışan bir SRY geni en az bir Y kromozomunun ortaya çıkmasıyla erkek olur. Bu nedenle XXYY kromozomuna sahip olan insanlar normalde erkek olur. XXYY sendromlu erkekler 46 yerine 48 kromozoma sahiptir. Bu yüzden XXYY sendromu bazen 48, XXYY sendromu olarak yazılır. Yaklaşık her 18.000-40.000 erkek doğumunda bir XXYY sendromlu bireyin doğduğu tahmin edilmektedir.
Diş hekimliğinde, hipodonti edinsel ya da doğumsal diş eksiklikleri olgusu için kullanılan terimlerdendir; anodonti ve oligodonti kavramları da hipodonti başlığı altında yer alan diş eksikliği olgularıdır.
Mikrognati (micrognathism), altçenenin yetersiz gelişmesi (hipoplazi) niteliğinde bir anomalidir; 1. ve 2. faringeal arklara (brankial yarık) özgü malformasyonların çoğu çene-yüz bölgesindeki mezenkimal dokunun embriyolojik dönemdeki yetersizliğine bağlanmaktadır. Altçene küçüktür ve geridedir; bu olgu, çevredeki kasların hareketlerini sınırlarken bu kaslardan yararlanan dokuların gelişmeleri de kısıtlanır. Kraniyofasiyal anomalileri içeren sendromlarda çok sık görülen bulgulardan biridir. Sendroma-özgü olmayan olguların varlığı da bilinmektedir; örneğin; altçenesi, kafa ile göğüs kafesi arasında sıkışmış olan bir fetüste çene kemiklerinin gelişimi olması gereken düzeye ulaşamamaktadır. Gebelik sorunları, gebelerin alkol kullanması, gestasyonel diabet (gebelik diabeti) gibi çevresel faktörler etkili olabilmektedir (Möbius sendromu). Olguların bir bömümü, gebelerdeki rubella (kızamıkçık) infeksiyonu komplikasyonudur. Yenidoğanların bir bölümünde görülebilen hafif mikrognatiler, altçene gelişiminin tamalanmasıyla silinebilir. Mikrognati'de altçene gövdesi kadar çene eklemini oluşturan yapıların hipoplazisi de önemlidir. Mikrognatilerin bir bölümü kulak sistemi, üstçene ve damak gelişiminin de aksadığı olgularla birlikte görülür.
Palilali, hecelerin, kelimelerin veya cümlelerin istem dışı tekrarı ile karakterize edilen bir dil bozukluğudur. Ekolali veya koprolali gibi diğer karmaşık tiklere benzeyen özelliklere sahiptir, ancak diğer afazilerden farklı olarak palilali, bağlamsal olarak doğru konuşmaya dayanır.
Mitokondriyal hastalık ya da mitokondri hastalıklıkları, insanlarda kalıtım yoluyla aktarılan ya da sonradan edinilen, mitokondri hasarının sonucunda ortaya çıkan ve bu hasar nedeniyle hücrenin enerji üretim mekanizmasını olumsuz etkileyen, genetik heterojenitenin görüldüğü hastalık grubudur.
Uniparental dizomi bir kromozomun iki kopyasının tek ebeveynden alındığı dizomik hücre dizileridir. Heterodizomi ve izodizomi olmak üzere iki sınıfa ayrılır. Bir ebeveynin her iki homolog kromozomu varsa heterodizomi adını alır. Aynı kromozomun iki kopyasını da alırsa buna izodizomi denir. Uniparental dizomi Silver Russel Sendromu, Angelman sendromu ve Prader Willi sendromuna neden olabilir.