
Down sendromu, trizomi 21 ya da mongolizm; genetik düzensizlik sonucu insanın 21. kromozom çiftinde fazladan bir kromozom bulunması durumu ve bunun sonucu olarak ortaya çıkan genetik bir bozukluktur. Down sendromu, bireyin 1 yaşından daha uzun süre yaşayabildiği tek otozomal trizomidir.

Klinefelter sendromu ya da 47, XXY sendromu; hücre bölünmesi sırasında, eşeysel kromozom düzensizliklerinden kaynaklanan semptomların kişide görülmesi durumudur.
Angelman sendromu ilk olarak 1965 yılında İngiliz doktor Harry Angelman tarafından tanımlanmış nörogenetik bir bozukluktur. Irklarda görülme hızı çok iyi bilinmemekle beraber yaklaşık ensidansın 15,000 ila 30,000 canlı doğumda bir olduğu kabul edilmektedir. Anneden gelen kromozom 15'teki bir bozukluktan kaynaklandığı sanılmaktadır. Hastalığın temel bulguları zeka geriliği, yürüyüş-koordinasyon bozukluğu, konuşma bozukluğu, konvülsiyon ve uygunsuz gülümsemelerdir. Hatta bu sebeple hastalık bazen “mutlu kukla ” sendromu olarak da bilinir.

Hiperdonti, artı dişlere, sürnümerer dişlere veya süpernümerer dişlere sahip olma veya başka bir deyişle olağandan fazla sayıda diş görünmesi durumudur. Diş arkının herhangi bir bölgesinde görülebilirler ve herhangi bir dişsel organı etkileyebilirler.
Ağız-Yüz-Parmak sendromu tip 1 , ektodermal displazi bulguları da içeren, X-kromozomu aracılığıyla dominant (XLD) geçen kalıtsal bir sendromdur. Simpson-Golabi-Behmel sendromu tip 2 ile alelik bağı olduğu belirlenmiştir. Kız çocuklarında görece sıktır. Erkek fetüsler, kalp ve beyin anomalilerinin neden olduğu intauterin ölümler nedeniyle kaybedilirler. Belirgin bir genel gelişme geriliği saptanır.
Oral-Facial-Digital sendrom II (orofaciodigital sendrom 2, Mohr sendromu; Mohr-Claussen sendromu), ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur. Belirgin bir genel gelişme geriliği saptanır.
Ağız-Yüz-Parmak sendromu tip 4 , ektodermal displazi bulguları da içeren, otosomal resesif geçen, ender görülen kalıtsal bir sendromdur. Belirgin bir genel gelişme geriliği saptanır.
OPD sendromu (otopalatodigital sendrom), X-kromozomu aracılığıyla dominant (XLD) yolla geçen kalıtsal bir sendromdur (Gorlin-Cohen sendromu hariç). Kız çocukları daha hafif etkilenir. Tümü benzer bulgular içeren 5 tipi vardır:
- Otopalatodigital sendrom tip I (OPD tip1)
- Otopalatodigital sendrom tip II (OPD tip2)
- Gorlin-Cohen sendromu (ilgili sayfaya gidiniz)
- Melnick-Needles sendromu (tip 1 ile allelik bağlantısı vardır; ilgili sayfaya gidiniz)
- Terminal osseöz displazi ve pigmentli deri defektleri (çok enderdir, ayrıntı verilmemiştir).
Gorlin-Cohen sendromu , ektodermal displazi bulgularını da içerebilen bir OPD sendromu tipidir. 2 fenotipi vardır:
- Frontometaphyseal dysplasia 1
- Frontometaphyseal dysplasia 2
Melnick-Needles sendromu , X-kromozomu aracılığıyla dominant (XLD) yolla geçen kalıtsal bir sendromdur. Kız çocukları daha hafif etkilenir. Erkek çocuklarında güçlü OPD tip 2 bulguları saptanır; çoğu ölü doğar. Gelişme geriliği olabilir. Başlıca bulgular şunlardır:
Mikrognati (micrognathism), altçenenin yetersiz gelişmesi (hipoplazi) niteliğinde bir anomalidir; 1. ve 2. faringeal arklara (brankial yarık) özgü malformasyonların çoğu çene-yüz bölgesindeki mezenkimal dokunun embriyolojik dönemdeki yetersizliğine bağlanmaktadır. Altçene küçüktür ve geridedir; bu olgu, çevredeki kasların hareketlerini sınırlarken bu kaslardan yararlanan dokuların gelişmeleri de kısıtlanır. Kraniyofasiyal anomalileri içeren sendromlarda çok sık görülen bulgulardan biridir. Sendroma-özgü olmayan olguların varlığı da bilinmektedir; örneğin; altçenesi, kafa ile göğüs kafesi arasında sıkışmış olan bir fetüste çene kemiklerinin gelişimi olması gereken düzeye ulaşamamaktadır. Gebelik sorunları, gebelerin alkol kullanması, gestasyonel diabet (gebelik diabeti) gibi çevresel faktörler etkili olabilmektedir (Möbius sendromu). Olguların bir bömümü, gebelerdeki rubella (kızamıkçık) infeksiyonu komplikasyonudur. Yenidoğanların bir bölümünde görülebilen hafif mikrognatiler, altçene gelişiminin tamalanmasıyla silinebilir. Mikrognati'de altçene gövdesi kadar çene eklemini oluşturan yapıların hipoplazisi de önemlidir. Mikrognatilerin bir bölümü kulak sistemi, üstçene ve damak gelişiminin de aksadığı olgularla birlikte görülür.
Kraniyofasiyal malformasyonlar ya da kraniyofasiyal anomaliler, baş-boyun ve yüz bölgesinin oluşma ve gelişme aşamalarındaki aksamalar ya da sapmalar sonucu ortaya çıkan yapısal ve işlevsel bozukluklardır. Genetik bilimindeki önemli ataklar, tüm kalıtsal hastalıklarda olduğu gibi kraniyofasiyal malformasyonlarda da tanı ve tedavi konusunda önemli gelişmelere yol açmıştır. Ancak, ender görülen sendromların ve fenotiplerinin çokluğu, kimi sendromların birbirleriyle çakışmaları uzmanların çabalarını güçleştirmektedir.
Kraniyosinostoz, kraniyosinostozis (craniosynostosis), kraniyofasiyal malformasyonların ve maksillofasiyal sendromların önemli bir bölümünde etkileri görülebilen konjenital bir patolojidir. Bu olgudaki temel bulgu kafatası eklemlerinin erken kapanmasıdır; etkilediği anatomik bölgelerde ortaya çıkan malformasyonlar, hangi suturaların ne düzeyde kapanmış olmasıyla orantılıdır. Malformasyonlar genellikle etkilenen eklemin dikey yönünde belirgindir.

Kraniyofasiyal yarıklar, kraniyofasiyal malformasyonların en önemlilerinden biridir; baş-boyun ve yüz bölgesinin oluşma ve gelişme aşamalarındaki aksamalar ya da sapmalar sonucu ortaya çıkan yapısal ve işlevsel bozuklukların önemli bir bölümünü oluştururlar. Embriyolojik kökenlerine göre; nöral tüp kökenli anomaliler, 1. ve 2. farengeal ark kökenli malformasyonlar, ektodermal displaziler söz konusudur.

Apert sendromu (acrocephalosyndactyly), otosomal dominant yolla aktarılan kalıtsal bir sendromdur. FGFR2 geni mutasyonuyla ortaya çıkan sendromlar kümesi üyelerinden biridir. FGFR2 geni, fibroblast growth factor reseptörüdür.

Mandibulofacial dysostosis sendromu, yüz bulgularının ön planda olduğu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. 1. ve 2. farengeal arklardaki gelişme bozukluğunun sonucudur. "Mikrosefali içeren tipi " ve "Alopesi" içeren tipi önemlidir. “Bauru tipi” çok enderdir. “Ptozis" içeren tip ise kaynaklarda bildirilmiş tek olgu olarak görülmektedir. Treacher Collins sendromu ile yakınlığı tartışılmaktadır.

Kleeblattschadel, kafatasında çok sayıda eklemin erken kapanması (kraniyosinstoz) sonucunda ortaya çıkan konjenital bir malformasyondur. Büyük bölümünün nedeni bilinmemektedir; bir bölümü sendroma özgüdür.
Larsen sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Ana nitelikleri nedeniyle, osteokondrodisplazi ve OPD sendromu 1 ile benzerlikleri vardır. Kalça, dirsek, bilek, diz gibi büyük eklemlerin çıkıkları, ayak deformasyonları, özellikle el başparmaklarda belirgin olan yassılaşma, skolyoz, boyun omurlarının malformasyonlarından kökenli kifoz, spina bifida, iskelet sistemiyle ilgili başlıca bulgulardır. Ayrıca, fibula, iki tarafta da tibianın önündedir (dislokasyon). El ve ayak parmaklarında çeşitli anomalilere rastlanabilir. Tırnaklar kısa ve küçüktür.

Myhre sendromu (LAPS sendromu), çoğu sporadik olarak görülen bir sendromdur; otosomal dominant yolla aktarılan olgular da bildirilmiştir.
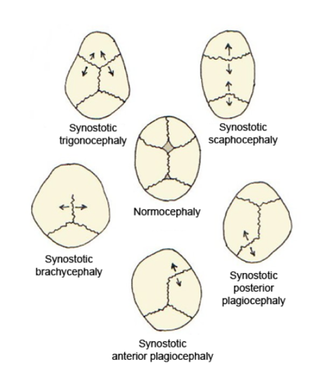
Brakisefali, türü için tipik olandan daha kısa bir kafatası şeklidir. Bazı evcil köpek ve kedi ırklarında, özellikle pug ve İran ırkında istenilen bir özellik olarak algılanır ve diğer hayvan türlerinde normal veya anormal olabilir. İnsanlarda, sefalik bozukluk düz kafa sendromu olarak bilinir ve koronal sütürlerin erken füzyonundan veya dış deformasyondan kaynaklanır. Koronal sütür, frontal kemiği kafatasının iki parietal kemiğiyle birleştiren fibröz eklemdir. Parietal kemikler kafatasının üstünü ve yanlarını oluşturur. Bu özellik Down sendromunda görülebilir.