
Sturge-Weber sendromu, ender görülen, gen mutasyonuna bağlı konjenital bir sendromdur. Kafatası normalden büyüktür (makrosefali).Gözlerden biri olmayabilir. Göz içindeki koroid pleksus adı verilen yapı içinde oluşan hemangioma nedeniyle göz içi sıvısı ve basıncı artmıştır. Yüzde porto şarabı renginde damar genişlemelerine bağlı geniş lekeler görülür. Yüzde, N.ophthalmicus ve N.trigeminalis olarak adlandırılan göz ve yüz sinirleri boyunca oluşan nevus flammeus'lar görülür; ağzın bir tarafında, dudak ve yanak mukozası ile dilde damar hiperplazisi saptanır. Dildeki damar artışı, etkilene bölgenin büyümesine yol açar. Dişeti büyümeleri, damarların artmasına ve epileptik hastalardaki dilantin tedavisine bağlıdır. Hemangiomun bulunduğu çene yarısındaki dişlerde hızlı gelişme ve hızlı sürme bulguları saptanır; bu bölgeden yapılan diş çekimlerinde damar zenginliği nedeniyle aşırı kanama olur.
Ektodermal displazi sendromları, ektodermden gelişen doku ve organlardaki malformasyonların ve oluşum kusurlarının saptandığı geniş bir topluluktur. Dişlerle ilgili anomaliler ve malformasyonlar; saç oluşumunda yetersizlikler (seyrek/kırılgan/oluşmama); ter bezlerinin eksikliğine bağlı terleme azlığı/yokluğu (hipohidroz); başkaca çeşitli deri ve tırnak patolojileri ortak bulgular olarak saptanır. Günümüze dek ektodermal displazi bulgularını içeren 200'e yakın sendrom bildirilmiştir; bu kadar çok sayıda fenotip bulunmasının nedeni farklı genlerde oluşan mutasyonlardır. Ektodermal displazili ailelerdeki kalıtım otosomal dominant, otosomal resesif ya da x-kromozomu aracılığıyla resesif olarak aktarılır.
Clouston sendromu, ektodermal displazi bulguları içeren, otosomal dominant geçen kalıtsal bir sendromdur. Ter bezlerinin ve dişlerin oluşumuyla ilgili sorunlar görülmez. Ancak, fiziksel ve zeka geriliği bulgularına nörolojik bulgular da eşlik eder. Görme kusurları saptanabilir.
KID sendromu (keratitis-ichthyosis-deafness), ektodermal displazi ve sağırlık bulgularını bulgularını içeren, otosomal dominant yolla aktarılan kalıtsal bir sendromdur.
Kohlschütter-Tönz sendromu, ektodermal displazi bulguları da içerebilen, otosomal resesif yolla geçen kalıtsal bir nörolojik sendromdur. Hastalarda, belirgin bir psikomotor gerilik saptanır; bu bulgunun temel nedeni, beyin ve beyincik (cerebellum) hipoplazisidir. Beyin boşlukları (ventriküller) geniştir. Elektroensefalografi (EEG)’de yoğun ve karmaşık elektriksel aktivite (hypsarrhythmia) bulunur. Zamanla azalabilen epileptik ataklar ve epilepsiye bağlı beyin zararları (ensefalopati) izlenir. Kas tonuslarında artma (hipertoni), spastik yapı ve ataksi görülür. Konuşma ve iletişim güçlüğü çekerler. Tüm bu bulgulara güçlü bir zeka geriliği eşlik eder.
Andersen-Tawil sendromu (Andersen sendromu), otosomal dominant yolla aktarılan kalıtsal bir sendromdur; 20 fenotipi vardır.
Au-Kline sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; böbrek, kalp ve yüz bulgularıyla öne çıkar.
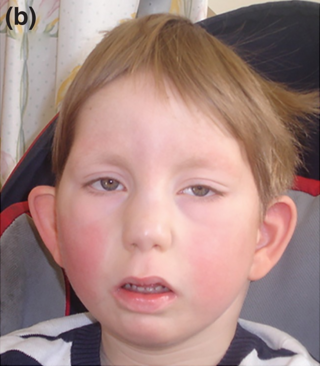
Galloway-Mowat sendromu, kalıtsal bir sendromdur. 8 fenotipinin 7'si otosomal resesif yolla aktarılır. Genel gelişme geriliği nedeniyle boy kısadır. Mikrosefali ve küçük bir yüz yapısı saptanır; kafa yanlardan basıktır, alın bombesi yüksektir. Fenotiplerin bir bölümün saptanan çok sayıdaki göz anomalisi nedeniyle görme kusurları belirlenir. Dış kulak ve burun malformasyonları olabilir. Altçene küçüktür (mikrognati), ağız açıklığı malformasyonları ve çukur damak görülebilir. Ağız bulgularının bir bölümü fenotipe özgüdür: mikrodonti, yarık damak, mine hipoplazisi saptanır. Fenotip 4 ve 5'te oral bulgu yoktur.

Kabuki sendromu, iki fenotipi olan bir sendromdur: Kabuki sendromu 1 ve Kabuki sendromu 2 (Kabuk2). Kabuk1, otosomal dominant yolla aktarılan kalıtsal ya da izole olgular biçiminde görülür; Kabuk2 ise, X-kromozomu aracılığıyla dominant yolla (XLD) aktarılır. Her iki fenotipini çok sayıda ortak bulguları vardır; özellikle yüz, iskelet sistemi, gelişme geriliği, parmakizleriyle ilgili bulgular ve zeka geriliği önemlidir.
MCAHS sendromu, üç fenotipten oluşan kalıtsal bir sendromdur; fenotip 1 (MCAHS1) ve fenotip 3 (MCAHS3) otosomal resesif yolla, fenotip 2 (MCAHS2) X-kromozomu aracılığıyla resesif (XLR) olarak aktarılır. Birbirlerinden çok farklı bulgular içeren MCAHS sendromlarında, çok sayıda malformasyonun yanı sıra özellikle sinir sistemini ilgilendiren bulgular öne çıkar.

Cowden sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. PTEN hamartoma tümör sendromları kümesi üyelerindendir. Bazı kaynakların listelerinde, PTEN kümesi sendromlarının tümü Cowden sendromu başlığı altında toplanmaktadır.

Proteus sendromu, AKT1 geni somatik mutasyonu ile ortaya çıkar. Bazı kaynaklarda, PTEN hamartoma tümör sendromları kümesi üyelerinden biri olarak nitelendirilir ya da Cowden sendromu başlığı altında toplanan sendrom grubu içinde sayılır. Son yıllarda, aşırı büyüme sendromlarından biri olarak değerlendirilmektedir.

Bannayan-Riley-Ruvalcaba sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. PTEN hamartoma tümör sendromları kümesi üyelerindendir. Bazı kaynakların listelerinde, PTEN kümesi sendromlarının tümü Cowden sendromu başlığı altında toplanmaktadır. Son yıllarda, aşırı büyüme sendromlarından biri olarak değerlendirilmektedir.
Klippel-Trenaunay sendromu, çok büyük bölümü nedeni saptanamayan izole bir sendrom olarak ortaya çıkar. Aşırı büyüme sendromları arasında gösterilmektedir.
Marshall-Smith sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Sotos sendromunun fenotipi sayılan Malan sendromuna genetik temelde kardeş (allelik) bir sendrom olarak nitelendiren uzmanlar vardır.

Multipl pterygium sendromu (Escobar sendromu), otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Lethal türünde fetüsün yaşama yetisi yoktur.
Seckel sendromu, 9 fenotipi olan, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Doğumdan önce belirlenen ve yaşam boyu süren, cücelik düzeyinde gelişme geriliği ile kuş kafasını andıran yüz görünümü, mikrosefali ve zeka geriliği saptanır. Alın dardır ve geriye çekilmiş gibidir. Gözler fırlaktır; iki taraflı optik atrofi nedeniyle görme sorunları yaşarlar; gözler çekiktir, kapaklarında yapışıklıklar vardır. Stabismus olabilir. Burun kuş gagasını çağrıştırır. Kulaklar biçimsizdir, kulak memesi yoktur ve işitme güçlüğü belirlenir. Asimetrik bir yüzde, altçene küçüktür ve geridedir (mikroretrognati), damak çukurdur ve yarık saptanır. Dişler küçüktür (mikrodonti) ve eksiktir (hipodonti); bu nedenlerle, çiğneme düzlemi bozuktur (maloklüzyon). Mine hipoplazisi belirgindir.

Stromme sendromu otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Mikrosefalinin yanı sıra göz, sindirim sistemi, kardiyovasküler sistem ve üriner sistem malformasyonları vardır. Bebek ölümleri sık görülür.
Shprintzen-Goldberg sendromu , marfanoid yapı, kraniyosinostoz, kardiyovasküler ve nörolojik anomalilerle karakterize, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Marfanoid yapı, Marfan sendromundaki bulguların büyük bölümünün saptandığı, ancak Marfan sendromunudaki genetik altyapının bulunmadığı olgular için kullanılan nitelemedir; uzun kollar ve bacaklar, örümceksi parmaklar (araknodaktili), eklemlerde aşırı gevşeklik ve mitral kapak prolapsusu Marfan sendromunu andıran başlıca bulgulardır. Robbins Shprintzen-Goldberg sendromu’nun, Marfan sendromu ve Loeys-Dietz sendromu ile oldukça çok büyük benzerlikleri vardır.

Limb-mammary sendromu, ektodermal displazi bulguları, el-ayak anomalileri ve memelerdeki oluşum kusurlarıyla karakterize otosomal dominant yolla aktarılan kalıtsal bir sendromdur. TP63 geninin mutasyonu sonucu ortaya çıkan sendromlardandır.
