
Organik kimya, organik bileşiklerin ve organik maddelerin yani karbon atomlarını içeren çeşitli formlardaki maddelerin yapısını, özelliklerini ve reaksiyonların bilimsel çalışmasını içeren, kimyanın bir alt dalıdır. Yapının incelenmesi yapısal formüllerini belirler. Özelliklerin incelenmesi, fiziksel ve kimyasal özellikleri ve davranışlarını anlamak için kimyasal reaktivitenin değerlendirilmesidir. Organik reaksiyonların incelenmesi doğal ürünlerin, ilaçların ve polimerlerin kimyasal sentezini ve bireysel organik moleküllerin laboratuvarda ve teorik çalışma yoluyla incelenmesidir.
Yoğun madde fiziği, maddenin yoğun hallerinin fiziksel özellikleriyle ilgilenen bir fizik dalıdır. Yoğun madde fizikçileri bu hallerin davranışını fizik kurallarını kullanarak anlamaya çalışır. Bunlar özellikle kuantum mekaniği kuralları, elektromanyetizma ve istatistiksel mekaniği içerir. En bilinen yoğun fazlar katı ve sıvılardır, harici yoğun fazlar ise düşük sıcaklıktaki bazı materyaller tarafından gösterilen üstünileten faz, atom kafeslerindeki dönüşlerin ferromanyetik ve antiferromanyetik fazları ve soğuk atom sistemlerinde bulunan Bose-Einstein yoğunlaşması. Araştırma için uygun sistemlerin ve fenomenlerin çeşitliliği yoğun madde fiziğini modern fiziğinin en aktif alanı yapıyor. Her 3 Amerikan fizikçiden biri kendini yoğun madde fizikçisi olarak tanımlıyor ve Yoğun Madde Fiziği Bölümü Amerikan Fizik Topluluğu’ndaki en geniş bölümdür. Bu alan kimya, malzeme bilimi ve nano teknoloji ile örtüşür ve atom fiziği ve biyofizikle de yakından ilgilidir. Teorik yoğun madde fiziği teorik parçacık ve nükleer fizikle önemli kavramlar paylaşır.
Kuantum Kimyası diğer adıyla Yeni Nicem, Kuantum mekaniğinin atom ve moleküllere uygulanması ile ilgilenen Kimya altdalıdır. Temel bir dal, yani saf Kimya olan Kuantum Mekaniği'nin bir uygulaması olduğundan uygulamalı Kimya dalı olarak değerlendirilebilir. Kuantum Kimyası'nda Schrödinger, Dirac, vb dalga denklemlerinin çözümüyle ilgilenilir. Ancak genellikle en çok tercih edilen, EM alan yokluğunda, spinsiz ve rölativistik olmayan Schrödinger denkleminin çözümüdür. Tek elektronlu sistemler dışında Schrödinger denklemi analitik olarak çözülemediğinden, çok elektronlu sistemler için nümerik çözümler yapılır. Kuantum Kimyası'nda bu nümerik çözümleri yapmak üzere çeşitli yöntemler vardır. Bunlar;
- Sıfırdan teorik yöntemler
- Yarı-ampirik yöntemler
- Yoğunluk fonksiyoneli yöntemleri
Deneysel verilerin kullanılmadığı, teorik ilkeler üzerine kurulu kuantum kimyasal hesaplamalara ab initio denir. Ab initio yönteminde genellikle karmaşık bir fonksiyonun daha basit fonksiyonlara indirgenmesi gibi matematiksel yaklaşımlar kullanılır.

Hesaplamalı fizik, fizik sorunlarını çözebilmek için sayısal algoritmaların üretilmesi ve gerçeklenmesini içerir. Genelde kuramsal fizikin bir alt dalı olarak değerlendirilir ancak bazen de kuramsal ve deneysel fizik arasında orta bir dal olarak da düşünülür.
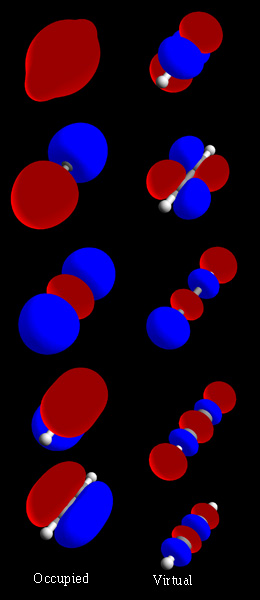
Kimyada moleküler orbital, bir molekül içindeki elektronların dalga benzeri davranışını tanımlayan matematiksel işlevdir. Molekülün herhangi bir bölümünde bir elektron bulma olasılığı gibi kimyasal ve fiziksel özellikleri hesaplamaya yarar.
Hesaplamalı kimya, kimya problemlerini çözmeye yardımcı olmak için bilgisayar simülasyonunu kullanan bir kimya dalıdır. Moleküllerin, katıların yapı ve özelliklerini hesaplamak için verimli bilgisayar programlarına dahil edilmiş teorik kimya yöntemlerini kullanır. Bu yöntemlerin kullanılmasının nedeni, hidrojen moleküler iyonu ile ilgili nispeten yeni sonuçlar dışında, kuantum çok-gövdeli(many-body) problemlerin analitik olarak çözülemez oluşudur. Hesaplama sonuçları normal olarak kimyasal deneylerle elde edilen bilgileri tamamlarken, bazı durumlarda gözlemlenmeyen kimyasal olayları da tahmin edebilmektedir. Yeni ilaç ve materyallerin tasarımında yaygın olarak kullanılmaktadır.

Bilimsel hesaplama karmaşık problemleri anlamak ve çözmek için gelişmiş bilgi işlem yeteneklerini kullanan çok disiplinli bir alandır. Hesaplamalı bilim üç farklı unsuru birleştirmektedir:
Kuantum kimyası bilgisayar programları, kuantum kimyası metodlarını uygulamak için bilgisayarlı kimyada kullanılır. Çoğu program, Hartree-Fock (HF) ve bazı post Hartree-Fock yöntemlerini içerir ve ayrıca yoğunluk fonksiyonları teorisi (DFT), moleküler mekanik veya yarı-ampirik kuantum kimyası metotlarını da içerebilirler. Bahsi geçen programlar arasında açık kaynaklı ve ticari yazılımlar bulunur. Bunların çoğu büyüktür, çoğu zaman birkaç ayrı program içerir ve uzun yıllar boyunca geliştirilmiştir.

Spartan, Wavefunction'ın moleküler modelleme ve bilgisayarlı kimya uygulamasıdır. Moleküler mekanik, yarı-ampirik yöntemler, ab initio modeller, yoğunluklu fonksiyonel modeller, post Hartree-Fock modeller, G3 (MP2) ve T1 içeren termokimyasal tarifler için kodlar içerir.
Katı hâl fiziğindeki hesaplamalı kimya yöntemleri, moleküller için uyguladıkları aynı yaklaşımı katı hâl fiziğinde de takip eder, ancak bu durumda iki farklılık gösterirler. Birincisi, katının translasyon simetrisinin kullanılması şarttır ve ikincisi, moleküler atom merkezli temel fonksiyonlara bir alternatif olarak düzlem dalgalar gibi tamamen dağılmış taban fonksiyonlarını kullanmak mümkündür. Bir kristalin elektronik yapısı genel olarak Brillouin kuşağındaki her nokta için elektron orbitallerinin enerjilerini tanımlayan bir bant yapısı ile tanımlanmaktadır. Ab initio ve yarı-ampirik hesaplamalar yörünge enerjilerini verir, bu nedenle bant yapı hesaplamalarına uygulanabilirler. Bir molekül için enerjiyi hesaplamak zaman alıcı olduğu için, Brillouin bölgesindeki tüm noktaların listesi için hesap yapmak daha da zaman alıcıdır.

Moleküler modelleme, moleküllerin davranışını modellemek veya taklit etmek için kullanılan teorik ve bilgisayarlı tüm yöntemleri kapsar. Bu yöntemler, küçük kimya sistemlerinden büyük biyolojik moleküllere ve malzeme gruplarına kadar değişen moleküler sistemleri incelemek için bilgisayarlı kimya, ilaç tasarımı, bilgisayarlı biyoloji ve malzeme bilimi alanlarında kullanılmaktadır. En basit hesaplamalar elle yapılabilir, ancak kaçınılmaz olarak makul büyüklükteki herhangi bir sistemin moleküler modellemesini bilgisayarların yapması gerekir. Moleküler modelleme yöntemlerinin ortak özelliği, moleküler sistemlerin atom düzeyinde tanımlanmasıdır. Bu, atomları en küçük bireysel birim olarak muamele edilmesini içerebilir veya protonları ve nötronları kuarkları, kuarkları, gluonlarıyla beraber ve elektronları da fotonlarıyla beraber açıkça modellemeyi içerebilir.
Jaguar hem gaz hem de çözelti fazları için ab initio kuantum kimya hesaplamalar yapmak amacıyla kullanılan bir bilgisayar yazılımı paketidir. Schrödinger şirketi tarafından pazarlanan ticari bir yazılımdır. Program, Richard Friesner ve William Goddard'ın araştırma grupları tarafından üretildi ve başlangıçta PS-GVB olarak adlandırıldı.
Gaussian ilk kez Gauss 70 adında 1970 yılında Carnegie Mellon Üniversitesi'ndeki John Pople ve araştırma grubu tarafından yayınlanan genel amaçlı bilgisayarlı kimya yazılım paketi. 1970'ten beri sürekli olarak güncellendi. Programın güncel sürümü Gaussian 16'dır. Başlangıçta Quantum Chemistry Program Exchange aracılığıyla yayınlanan program, daha sonra Carnegie Mellon Üniversitesi'ne lisanslanmıştır ve 1987'den beri Gaussian, Inc. tarafından geliştirilmiştir.

CP2K, katı hal, sıvı, moleküler ve biyolojik sistemlerin atomistik simülasyonlarını gerçekleştirmek için Fortran 2003'te yazılan serbestçe kullanılabilen (GPL) bir programdır.
NWChem, hem kuantum kimyasal ve hem de moleküler dinamik işlevselliği içeren ab initio bilgisayarlı kimya yazılımıdır. Geleneksel iş istasyonu kümelerinin yanı sıra yüksek performanslı paralel süper bilgisayarlarda da çalışacak şekilde tasarlanmıştır.

Moleküler mekanik moleküler sistemleri modellemek için klasik mekaniği kullanır. Born-Oppenheimer yaklaşımının geçerli olduğu varsayılır ve tüm sistemlerin potansiyel enerjisi, kuvvet alanları kullanılarak nükleer koordinatların bir fonksiyonu olarak hesaplanır. Moleküler mekanik, boyutu birkaç atom büyüklüğünde olan sistemlerden tutun da milyonlarca atomdan oluşan büyük sistemlere kadar uygulanabilir.
Teorik ve hesaplamalı kimyada temel seti, Hartree Fock yöntemi veya yoğunluk fonksiyonel teorisinde kullanılan bir dizi fonksiyondur. Temel setleri Hartree Fock yöntemi veya yoğunluk fonksiyonel teorisindeki elektronik dalga fonksiyonunu temsil etmeyi amaçlarlar. Modelin kısmi diferansiyel denklemlerinin bilgisayarda verimli uygulanması amacı ile uygun cebirsel denklemlere dönüştürülmesinde kullanılırlar.

SIESTA, moleküller ve katıların üzerine ab initio moleküler dinamik simülasyonlarını gerçekleştirmek ve etkilielektronik yapı hesaplamalarını yapabilmek için tasarlanmış orijinal bir yöntem ve bu yöntemin bilgisayar programı uygulamasıdır. SIESTA'nın verimliliği, kesinlikle yerelleştirilmiş temel kümelerin kullanılmasından ve uygun sistemlere uygulanabilen doğrusal ölçekleme algoritmalarının varlığından kaynaklanmaktadır. Kodun çok önemli bir özelliği, doğruluk ve maliyetinin, hızlı keşif hesaplamalarından- düzlem dalga ve tüm elektron yöntemleri gibi diğer yaklaşımların kalitesiyle eşleşen- son derece hassas simülasyonlara kadar geniş bir aralıkta ayarlanabilmesidir.
Atom fiziğinde, etkin nükleer yük, çok elektronlu bir atomda bir elektronun yaşadığı gerçek pozitif (nükleer) yük miktarıdır. "Etkili" terimi, negatif yüklü elektronların koruyucu etkisi, daha yüksek enerjili elektronların, iç katmanın itici etkisi nedeniyle çekirdeğin tam nükleer yükünü deneyimlemesini engellediği için kullanılır. Bir elektronun deneyimlediği etkin nükleer yüke çekirdek yükü de denir. Atomun oksidasyon sayısı ile nükleer yükün gücünü belirlemek mümkündür. Elementlerin fiziksel ve kimyasal özelliklerinin çoğu, elektronik konfigürasyon temelinde açıklanabilir. İyonlaşma enerjilerinin davranışını düşününperiyodik tabloda. İyonizasyon potansiyelinin büyüklüğünün aşağıdaki faktörlere bağlı olduğu bilinmektedir:
- atomun boyutu;
- nükleer yük;
- İç kabukların eleme etkisi ve
- En dıştaki elektronun, içteki elektron tarafından kurulan yük bulutuna nüfuz etme derecesi.