Oculofaciocardiodental sendrom
Oculofaciocardiodental sendrom, göz malformasyonları, çene-yüz anomalileri, dolaşım sistemi ve bağışıklık sistemi defektleriyle karakterize, X-kromozomu aracılığıyla dominant (XLD) yolla aktarılan kalıtsal bir sendromdur. Mikroftalmi sendromunun 18 fenotipinden biridir.[1][2]
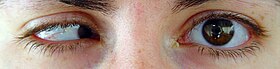
Genel gelişme geriliği saptanır. Mikrosefali bulgusu bazı hastalarda çok belirgindir. Yüz dar ve uzundur. Kaşlar kalın ve yay biçimindedir. Çok sayıda göz bulgusu vardır. Özellikle mikroftalmi (küçük göz) ya da kriptoftalmi (yok denecek kadar küçük göz) saptanır.[2][3][4][5]

Kornea küçüktür (mikrokornea). Konjenital katarakt, glokom ve dışa şaşılık (ekzotropi) bulguları vardır. Göz kapağı düşüklüğü (ptozis) ve göz kapaklarında yapışıklık olabilir. Burun sırtı belirgin, ucu iki lobludur. Kulaklar iri-asimetrik ve biçimsizdir, işitme sorunları vardır.[2][3]
Damak ve küçük dil yarıkları (uvula bifida) saptanır. Özellikle köpek dişlerinin kökleri kalındır. Dişlerde mine defektleri vardır. Çok sayıda diş eksikliği saptanır (oligodonti) ya da artı dişler (süpernümerer dişler) görülür. Dişlerdeki sürme aksamalarına bağlı çiğneme düzlemi deformasyonları (maloküzyon) oluşur.[2][3][6][7]
Kalpte atrial ve ventriküler septal defektler, mitral kapak prolapsusu, kalp kapaklarında darlık, triküspid kapakta yetmezlik oldukça önemli dolaşım sistemi malformasyonlarıdır.[2][3][6]
İskelet sisteminde kol kemiklerinin (radius ve ulna) kaynaşması, ayak parmaklarında yapışıklıklar (sindaktili) ve skolyoz bulguları vardır.[2][3][6] Psikomotor gerilik saptanır. Görme yolları (optik kiazma) gelişememiştir ve taslak halinde kalmıştır.[2][3]
Kaynakça
- ^ Hilton E, Johnston J, Whalen S, et al. BCOR analysis in patients with OFCD and Lenz microphthalmia syndromes, mental retardation with ocular anomalies, and cardiac laterality defects. European Journal of Human Genetics, 17: 1325-1335, 2009
- ^ a b c d e f g Gorlin RJ, Marashi AH, Obwegeser HL. Oculo-facio-cardio-dental (OFCD) syndrome. American Journal of Medical Genetics, 63: 290-292, 1996
- ^ a b c d e f Schulze BR, Horn D, Kobelt A, Tariverdian G, Stellzig A. Rare dental abnormalities seen in oculo-facio-cardio-dental (OFCD) syndrome: three new cases and review of nine patients. American Journal of Medical Genetics, 82(5):429-435, 1999
- ^ Oberoi S, Winder AE, Johnston J, et al. Case reports of oculofaciocardiodental syndrome with unusual dental findings. American Journal of Medical Genetics A, 136A: 275-277, 2005
- ^ Horn D, Chyrek M, Kleier S, et al. Novel mutations in BCOR in three patients with oculo-facio-cardio-dental syndrome, but none in Lenz microphthalmia syndrome. European Journal of Human Genetics, 13(5):563-569, 2005
- ^ a b c Tsukawaki H, Tsuji M, Kawamoto T, Ohyama K. Three cases of oculo-facio-cardio-dental (OFCD) syndrome. The Cleft Palate-Craniofacial Journal, 42(5):467-476, 2005
- ^ Lubinsky M, Kantaputra PN. Syndromes with supernumerary teeth. American Journal of Medical Genetics, 170A:2611–2616, 2016










