
Down sendromu, trizomi 21 ya da mongolizm; genetik düzensizlik sonucu insanın 21. kromozom çiftinde fazladan bir kromozom bulunması durumu ve bunun sonucu olarak ortaya çıkan genetik bir bozukluktur. Down sendromu, bireyin 1 yaşından daha uzun süre yaşayabildiği tek otozomal trizomidir.
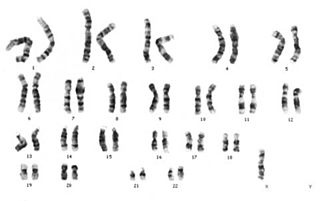
Turner sendromu, kromozom anomalisi sonucu ortaya çıkan ve kız çocuklarını etkileyen bir sendromdur; bir dişide eşey kromozomlarından birinin bulunmaması sonucu ortaya çıkar. Turner sendromluların fenotipi dişi olarak görülür fakat; eşey organları ve eşey hücreleri gelişmez. Kısır bireylerdir.

İnsan dişi, besinleri yutmaya ve sindirmeye hazırlık aşamasında keserek ve ezerek besinlerin mekanik olarak yıkımında görev yapar. İnsanlarda, her birinin belirli bir işlevinin olduğu kesici diş, köpek dişi, küçük azı dişi ve azı dişi olmak üzere dört tip diş vardır. Kesici dişler besini keser, köpek dişleri besini koparır ve küçük azı ve azı dişleri besini ezer. Dişlerin kökleri maksilla ya da mandibula içerisine yerleşmiş ve diş eti ile kaplanmıştır. Dişler yoğunluğu ve sertliği farklı çeşitli dokulardan yapılmıştır.

Kedi miyavlaması sendromu, Kedi çığlığı sendromu veya tıptaki isimleriyle Cri du Chat sendromu ya da Cri-du-Chat sendromu, 5. kromozomun bir parçasının kaybıyla ilişkili nadir bulunan bir genetik düzensizliktir. Sendromun genetik tanımı 45,X(X/Y),-5p olarak gösterilir. Yani kişide 45 kromozomun bulunduğunu fakat 5. kromozomun kısa (petit) kolunun bir kısmının bulunmadığını ifade eder. Bu tip kromozom mutasyonlarında DNA'daki bazın ya da bazların yok olmasına delesyon adı verilir. Delesyondaki büyüklük bebeklerdeki fiziksel, psikomotor ve zihinsel gelişimlerini etkiler.
Rett sendromu, yaygın gelişimsel bozukluklardan birisi olarak sınıflandırılan beyinsel gelişim bozukluğudur. Ancak bunun yanlış bir sınıflandırma olduğunu ve benzer şekilde otistik belirtiler gösteren frajil X sendromu, tüberoz skleroz ya da Down sendromunun yaygın gelişimsel bozukluklar olarak sınıflandırılabileceğini önesüren görüşler bulunmaktadır. Bu sendromun belirtileri kolaylıkla otizm ve Angelman sendromunun belirtileriyle karışır. Klinik belirtiler arasında baş büyüme hızının azalması ve bazen mikrosefali, küçük el ve ayaklar bulunur. Stereotipik ve yineleyici el hareketleri de gözlenir. Bilişsel bozukluk ve gerileme döneminde de sosyalleşme sorunları da belirtiler arasında görülür. Okula girdikleri dönemde sosyalleşme genellikle düzelir. Rett sendromu olan kız çocuklar gastrointestinal bozukluklara yakalanmaya yatkındır ve %80’i nöbet geçirir. Hemen hemen hiç sözel becerileri yoktur ve kadınların %50’si yürüyemez. Skolyoz, büyüme eksikliği ve kabızlık çok yaygındır ve sorunlu olabilir.

Köpek dişi, memeli oral anatomisinde incelenen uzun ve sivri diş. Fakat daha düz bir şekilde ortaya çıkabilirler. Bunun sonucu öndeki kesici dişlere benzerler. Öncelikli olarak sert yiyeceği parçalamakla görevlidirler. İkincil kullanımları ise saldırıya yöneliktir. Çoğunlukla bütün memelilerde en büyük dişlerdir. Çoğu memelide ikisi alt çenede ve ikisi üst çenede olmak üzere dört adet köpek dişi bulunur. Aynı çenede bulunan köpek dişlerini birbirinden, kesici dişler ayırır. Örneklerini köpeklerde ve insanlarda görebiliyoruz.

Ellis-van Creveld sendromu, ektodermal displazi bulguları içeren, otosomal resesif geçen kalıtsal bir sendromdur. Asfiksiyan torasik displazi sendromu'nun 22 fenotipinden 2'si Ellis-van Creveld sendromu olarak değerlendirilir.
Jadassohn-Lewandowsky sendromu, ektodermal displazi bulguları içeren, otosomal dominant geçen kalıtsal bir sendromdur.
Melnick-Needles sendromu , X-kromozomu aracılığıyla dominant (XLD) yolla geçen kalıtsal bir sendromdur. Kız çocukları daha hafif etkilenir. Erkek çocuklarında güçlü OPD tip 2 bulguları saptanır; çoğu ölü doğar. Gelişme geriliği olabilir. Başlıca bulgular şunlardır:
Mikrognati (micrognathism), altçenenin yetersiz gelişmesi (hipoplazi) niteliğinde bir anomalidir; 1. ve 2. faringeal arklara (brankial yarık) özgü malformasyonların çoğu çene-yüz bölgesindeki mezenkimal dokunun embriyolojik dönemdeki yetersizliğine bağlanmaktadır. Altçene küçüktür ve geridedir; bu olgu, çevredeki kasların hareketlerini sınırlarken bu kaslardan yararlanan dokuların gelişmeleri de kısıtlanır. Kraniyofasiyal anomalileri içeren sendromlarda çok sık görülen bulgulardan biridir. Sendroma-özgü olmayan olguların varlığı da bilinmektedir; örneğin; altçenesi, kafa ile göğüs kafesi arasında sıkışmış olan bir fetüste çene kemiklerinin gelişimi olması gereken düzeye ulaşamamaktadır. Gebelik sorunları, gebelerin alkol kullanması, gestasyonel diabet (gebelik diabeti) gibi çevresel faktörler etkili olabilmektedir (Möbius sendromu). Olguların bir bömümü, gebelerdeki rubella (kızamıkçık) infeksiyonu komplikasyonudur. Yenidoğanların bir bölümünde görülebilen hafif mikrognatiler, altçene gelişiminin tamalanmasıyla silinebilir. Mikrognati'de altçene gövdesi kadar çene eklemini oluşturan yapıların hipoplazisi de önemlidir. Mikrognatilerin bir bölümü kulak sistemi, üstçene ve damak gelişiminin de aksadığı olgularla birlikte görülür.
MCAHS sendromu, üç fenotipten oluşan kalıtsal bir sendromdur; fenotip 1 (MCAHS1) ve fenotip 3 (MCAHS3) otosomal resesif yolla, fenotip 2 (MCAHS2) X-kromozomu aracılığıyla resesif (XLR) olarak aktarılır. Birbirlerinden çok farklı bulgular içeren MCAHS sendromlarında, çok sayıda malformasyonun yanı sıra özellikle sinir sistemini ilgilendiren bulgular öne çıkar.

Goltz sendromu, X kromozomu aracılığıyla dominant (XLD) olarak aktarılan kalıtsal bir sendromdur; erkek fetüslerin çok büyük bölümü intrauterin evrede (rahimde) öldüğü için hastaların çoğu kız bebeklerdir.
Multipl konjenital anomaliler-hipotoni-epilepsi sendromu, 3 fenotipi olan kalıtsal bir sendromdur. Fenotip 1 (MCAHS1) ve fenotip 3 (MCAHS3) otosomal resesif, tip 2 (MCAHS2) ise x-kromozomu aracılığı ile resesif olarak aktarılır. Birçok enzimin çalışabilmesinde, kompleman sisteminin düzenlenmesinde ve hücrelerarası iletişimin sağlanmasında önemli katkıları olan, fosfogliserid yapısındaki glikosilfosfatidilinositol sentezindeki sorunlarından kökenlidir. Nörolojik belirtiler sendromun fenotiplerindeki temel bulguları oluştururlar; bunlara eklenen çok sayıdaki anomalilerin yeri, sayısı ve gücü farklıdır.
Pallister-Hall sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Hipotalamus'ta hamartoblastoma olarak nitelendirilen oluşumun yanı sıra çok sayıda endokrin sistem anomalileri, açılmamış anüs ve polidaktili bulguları ön plandadır. Hipofiz agenezi/hipoplazisi kökenli hipopituitarizm, adrenal gland hipoplazisi bulguları, hipotiroidizm, endokrin sistem anomalilerinin başlıcalarıdır.
Raine sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Yeni doğan ve bebek ölümleri sıktır; çoğu doğumdan sonraki haftalar içinde kaybedilen bebeklerde, kemiklerde yaygın yoğunlaşmalar (skleroz) saptanır. Özellikle kafa tabanı ve yüz kemiklerindeki skleroz oldukça belirgindir. Ayrıca, yoğun bir periostal kemik yapımı nedeniyle, uzun kemiklerin çevresinde sklerotik bir kılıf oluşur. Mikrosefali ve alın bombesi oldukça yüksek olan brakisefali (turribrakisefali) saptanır. Bıngıldaklar (fontaneller) tam kapanmamıştır. Kafatasındaki kemik yoğunlaşması (osteoskleroz) kraniyofasiyal displazi bulgularının oluşmasına yol açar. Ekzoftalminin yanı sıra hipertelorizm de olabilir. Kulaklar arkaya dönük ve biçimsizdir. Yüz orta bölümü hipoplazisi nedeniyle üstçene küçüktür (mikrognati), basık ve silik bir burun yapısı izlenir; bu yapı balık yüzü izlenimi verir. Altçene önde gibidir (prognatizm). Ağız açıklığı dardır. Derinliği fazla olan bir yarık damak varlığı beslenme bozukluklarına neden olur. Doğumda dişler olabilir, zamanında süren dişlerse küçüktür (mikrodonti). Hastaların büyük bölümünde dişeti büyümeleri vardır. Dil oldukça büyüktür (makroglossi).
Osteopathia striata, kemik yoğunlaşmasının (osteoskleroz) saptandığı, X-kromozomu aracılığıyla (XLD) aktarılan kalıtsal bir sendromdur; olguların 1/3’ü sporadiktir.

Meckel-Gruber sendromu, çok sayıda genin etkilenmesinden kaynaklanan, bu nedenle 15 fenotipi olan, otosomal resesif yolla aktarılan, siliyopatiler grubundan, kalıtsal bir sendromdur. Kistik böbrekler, beyin anomalileri ve ensefalosel, polidaktili ve karaciğer anomalileri başlıca bulgulardır. En sık görülen fenotip Meckel sendromu 1, öteki fenotiplerdeki bulguların çok büyük bir bölümünü içerir. Bazı fenotipleri Joubert sendromuyla çakışan nitelikleri içermektedir.
Elsahy-Waters sendromu (brachioskeletogenital sendrom), otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Kafa ve yüz bulguları, dişlerle ilgili patolojiler, vertebraların kaynaşması ile zeka geriliği temel bulguları arasındadır.
Meckel sendromu, birden fazla etkisi olan bir genle ilgili (pleiotropic), otosomal resesif yolla aktarılan, 15 kadar fenotipi bulunan kalıtsal bir sendromdur. Çok sayıda ve karmaşık bulguların varlığı tanıda güçlükler oluşturur. Bazı uzmanlar, Meckel sendromunun 3 temel bulgusu olduğunu savunurlar: (i) Santral sinir sistemi malformasyonları ; (ii) Kistik böbrekler; (iii) Karaciğer patolojileri. Bebeklerin yaşama yetisi çok kısıtlıdır; canlı doğabilenlerin çoğu birkaç hafta içimde yitirilir.
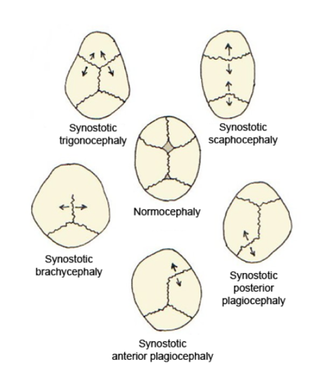
Brakisefali, türü için tipik olandan daha kısa bir kafatası şeklidir. Bazı evcil köpek ve kedi ırklarında, özellikle pug ve İran ırkında istenilen bir özellik olarak algılanır ve diğer hayvan türlerinde normal veya anormal olabilir. İnsanlarda, sefalik bozukluk düz kafa sendromu olarak bilinir ve koronal sütürlerin erken füzyonundan veya dış deformasyondan kaynaklanır. Koronal sütür, frontal kemiği kafatasının iki parietal kemiğiyle birleştiren fibröz eklemdir. Parietal kemikler kafatasının üstünü ve yanlarını oluşturur. Bu özellik Down sendromunda görülebilir.