Guillain-Barré sendromu (GBS), çevresel sinir sisteminin edinilmiş bir bağışıklık kökenli yangısal bozukluğudur; merkezi sinir sistemi etkilenmez. Bu hastalık için kullanılan diğer isimler şöyledir: akut enflamatuvar demiyelinize edici polinöropati, akut idiyopatik poliradikülonörit, akut idiyopatik polinörit, Fransız polyosu, Landry'nin yükselici felci.

Down sendromu, trizomi 21 ya da mongolizm; genetik düzensizlik sonucu insanın 21. kromozom çiftinde fazladan bir kromozom bulunması durumu ve bunun sonucu olarak ortaya çıkan genetik bir bozukluktur. Down sendromu, bireyin 1 yaşından daha uzun süre yaşayabildiği tek otozomal trizomidir.
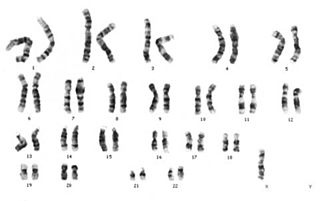
Turner sendromu, kromozom anomalisi sonucu ortaya çıkan ve kız çocuklarını etkileyen bir sendromdur; bir dişide eşey kromozomlarından birinin bulunmaması sonucu ortaya çıkar. Turner sendromluların fenotipi dişi olarak görülür fakat; eşey organları ve eşey hücreleri gelişmez. Kısır bireylerdir.

İnsan dişi, besinleri yutmaya ve sindirmeye hazırlık aşamasında keserek ve ezerek besinlerin mekanik olarak yıkımında görev yapar. İnsanlarda, her birinin belirli bir işlevinin olduğu kesici diş, köpek dişi, küçük azı dişi ve azı dişi olmak üzere dört tip diş vardır. Kesici dişler besini keser, köpek dişleri besini koparır ve küçük azı ve azı dişleri besini ezer. Dişlerin kökleri maksilla ya da mandibula içerisine yerleşmiş ve diş eti ile kaplanmıştır. Dişler yoğunluğu ve sertliği farklı çeşitli dokulardan yapılmıştır.

Kedi miyavlaması sendromu, Kedi çığlığı sendromu veya tıptaki isimleriyle Cri du Chat sendromu ya da Cri-du-Chat sendromu, 5. kromozomun bir parçasının kaybıyla ilişkili nadir bulunan bir genetik düzensizliktir. Sendromun genetik tanımı 45,X(X/Y),-5p olarak gösterilir. Yani kişide 45 kromozomun bulunduğunu fakat 5. kromozomun kısa (petit) kolunun bir kısmının bulunmadığını ifade eder. Bu tip kromozom mutasyonlarında DNA'daki bazın ya da bazların yok olmasına delesyon adı verilir. Delesyondaki büyüklük bebeklerdeki fiziksel, psikomotor ve zihinsel gelişimlerini etkiler.
Genetik bozukluk, genlerde ve kromozomlarda görülen anomaliler sonucu ortaya çıkan durumdur. Kanser gibi bazı hastalıklar yaşam sırasında edinilen ve bazı hücrelerde görülen genetik anomaliler nedeniyle olsa da "genetik hastalık" terimi genellikle vücuttaki tüm hücrelerde bulunan ve döllenmeden beri var olan hastalıklar için kullanılır. Bazı genetik bozukluklar, sperm ve yumurtalar gibi üreme hücrelerini oluşturan mayoz bölünme sırasında oluşan kromozom anomalileri nedeniyle ortaya çıkar. Bunlara örnek olarak Down sendromu, Turner sendromu (45X0) ve Klinefelter sendromu sayılabilir. Diğer genetik değişiklikler ebeveynler tarafından tohum hücrelerin oluşturulması sırasında ortaya çıkabilir. Bunlara bir örnek frajil X sendromu ya da Huntington hastalığına neden olabilen üçlü yayılma tekrar mutasyonudur. Hatalı genler ebeveynlerden olduğu gibi alınmış da olabilir. Bu genellikle sağlıklı ama resesif gen taşıyan iki kişinin üremesi ya da hatalı genin dominant olması sonucunda olabilir.
Rett sendromu, yaygın gelişimsel bozukluklardan birisi olarak sınıflandırılan beyinsel gelişim bozukluğudur. Ancak bunun yanlış bir sınıflandırma olduğunu ve benzer şekilde otistik belirtiler gösteren frajil X sendromu, tüberoz skleroz ya da Down sendromunun yaygın gelişimsel bozukluklar olarak sınıflandırılabileceğini önesüren görüşler bulunmaktadır. Bu sendromun belirtileri kolaylıkla otizm ve Angelman sendromunun belirtileriyle karışır. Klinik belirtiler arasında baş büyüme hızının azalması ve bazen mikrosefali, küçük el ve ayaklar bulunur. Stereotipik ve yineleyici el hareketleri de gözlenir. Bilişsel bozukluk ve gerileme döneminde de sosyalleşme sorunları da belirtiler arasında görülür. Okula girdikleri dönemde sosyalleşme genellikle düzelir. Rett sendromu olan kız çocuklar gastrointestinal bozukluklara yakalanmaya yatkındır ve %80’i nöbet geçirir. Hemen hemen hiç sözel becerileri yoktur ve kadınların %50’si yürüyemez. Skolyoz, büyüme eksikliği ve kabızlık çok yaygındır ve sorunlu olabilir.

Diabet ya da Diabetes mellitus, sıklıkla yalnızca diabet ya da diyabet veya halk arasında şeker hastalığı olarak adlandırılan, genellikle kalıtımsal ve çevresel etkenlerin birleşimi ile oluşan ve kandaki glukoz seviyesinin aşırı derecede yükselmesiyle (hiperglisemi) sonuçlanan metabolik bir bozukluktur. Vücutta kan şekerinin düzenlenmesi pek çok sayıda kimyasal madde ve hormonun karmaşık etkileşimi sonucunda sağlanır. Şeker metabolizmasının düzenlenmesinde rol oynayan hormonlardan en önemlisi pankreasın beta hücrelerinden salgılanan insülin hormonudur. Diyabetes Mellitus ya insülin salgılanmasındaki yetersizlik ya da insülinin etkisindeki veya insülin cevabındaki bir bozukluk sonucunda ortaya çıkan yüksek kan şekerinin yol açtığı birkaç grup hastalığı tanımlamak için kullanılan ortak bir terimdir.
Hipoplazi ya da az gelişim, bir organın yetersiz gelişme nedeniyle doğumsal olarak küçük kalmasıdır. Organ, tüm anatomik özelliklerini taşır, fizyolojik işlevlerini yapabilir. Körelme, irileşim ve aşırı gelişim az gelişimin (hipoplazi) karşıtı olan kavramlardır; bu olguların tümü edinseldir. Hipoplazi kavramına en yakın olan olgu aplazi olgusudur; hipoplazi ve aplazi doğumsal (konjenital) patolojilerdir.
Anatomide heterokromi, farklı renklerin bulunmasıdır. Genellikle iriste görülür ama bazen saçta ve deride de görülebilir. Heterokromi melanin (pigment) fazlalığının ya da eksikliğinin sonucudur. Bu kalıtsal ya da genetik mozaizm, kimerizm, hastalık, travma gibi nedenlerden dolayı olabilir.
Diş hekimliğinde, hipodonti edinsel ya da doğumsal diş eksiklikleri olgusu için kullanılan terimlerdendir; anodonti ve oligodonti kavramları da hipodonti başlığı altında yer alan diş eksikliği olgularıdır.
Kalıcı dişler ya da yetişkin dişleri difiyodont memelilerde oluşan ikinci diş takımıdır. İnsanlarda ve eski dünya maymunlarında, altı maksiller ve mandibular azı dişleri, dört maksiller ve mandibular küçük azı dişleri, iki maksiller ve mandibular köpek dişleri, dört maksiller ve mandibular kesici dişler olmak üzere toplamda otuz iki adet kalıcı diş vardır.
Ağız-Yüz-Parmak sendromu , günümüze dek 16 fenotipi belirlenmiş olan bir sendromlar kümesidir. Bilinen fenotiplere yenileri eklenebilir. Ağız-Yüz-Parmak sendromunun OFD arasında en sık ratlanılanı OFP tip I temel bulguları içerir; sıkça rastlanan öteki fenotiplerde, tip I'e eklenen ya da çıkarılan yan bulgular vardır.
Pierre Robin sequence ya da Pierre-Robin sequence , üç komponentli bir kraniyofasiyal anomalidir, Pierre Robin sequence, Pierre Robin sendromu'nun ana bulgusudur:
- Altçene hipoplazisi: Mandibular mikrognati, bu tablonun ana komponentidir. Altçene gelişemeyince dil gelişmesi de yetersiz kalır ve dil geride kalır; üst solunum yollarını kapatabilir.
- Glossoptosis: Dilin arkaya-geriye çekilmiş olmasıdır; dil, ağız boşluğunu tam olarak dolduramaz.
- Damak yarığı: Dilin ağız boşluğunu tam olarak dolduramaması damak kemiklerinin kaynaşmasını olumsuz etkiler – yetersiz indüksiyon mekanizması(*). Damak yarığının büyüklüğü glossoptosis bulgusunun gücüyle bağlantılıdır; glossoptosis'in hafif olduğu olgularda damak yarığı görülmeyebilir.
Mikrognati (micrognathism), altçenenin yetersiz gelişmesi (hipoplazi) niteliğinde bir anomalidir; 1. ve 2. faringeal arklara (brankial yarık) özgü malformasyonların çoğu çene-yüz bölgesindeki mezenkimal dokunun embriyolojik dönemdeki yetersizliğine bağlanmaktadır. Altçene küçüktür ve geridedir; bu olgu, çevredeki kasların hareketlerini sınırlarken bu kaslardan yararlanan dokuların gelişmeleri de kısıtlanır. Kraniyofasiyal anomalileri içeren sendromlarda çok sık görülen bulgulardan biridir. Sendroma-özgü olmayan olguların varlığı da bilinmektedir; örneğin; altçenesi, kafa ile göğüs kafesi arasında sıkışmış olan bir fetüste çene kemiklerinin gelişimi olması gereken düzeye ulaşamamaktadır. Gebelik sorunları, gebelerin alkol kullanması, gestasyonel diabet (gebelik diabeti) gibi çevresel faktörler etkili olabilmektedir (Möbius sendromu). Olguların bir bömümü, gebelerdeki rubella (kızamıkçık) infeksiyonu komplikasyonudur. Yenidoğanların bir bölümünde görülebilen hafif mikrognatiler, altçene gelişiminin tamalanmasıyla silinebilir. Mikrognati'de altçene gövdesi kadar çene eklemini oluşturan yapıların hipoplazisi de önemlidir. Mikrognatilerin bir bölümü kulak sistemi, üstçene ve damak gelişiminin de aksadığı olgularla birlikte görülür.

Kraniyofasiyal yarıklar, kraniyofasiyal malformasyonların en önemlilerinden biridir; baş-boyun ve yüz bölgesinin oluşma ve gelişme aşamalarındaki aksamalar ya da sapmalar sonucu ortaya çıkan yapısal ve işlevsel bozuklukların önemli bir bölümünü oluştururlar. Embriyolojik kökenlerine göre; nöral tüp kökenli anomaliler, 1. ve 2. farengeal ark kökenli malformasyonlar, ektodermal displaziler söz konusudur.

Dişeti büyümeleri, dişler arasında yer alan piramit biçimindeki dişetlerinin büyümesidir.

Russell-Silver sendromu, genel gelişme geriliğinin neden olduğu boy kısalığı ile yüz ve vücut asimetrisi saptanan; genetik kökeni karmaşık olan, ancak 5 fenotipinden 4’ünün otosomal dominant yolla aktarıldığı belirlenen bir sendromdur. Temel nedenin 11p15 (ICR1) distal kromozomunun yetersiz metilasyonu (hipometilasyon) ise olduğu gösterilmiş; aynı kromozomun hipermetilasyonunun ise Beckwith-Wiedemann sendromuna yol açabileceği ileri sürülmüştür.

Akromelik frontonazal disostoz, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Frontonazal displazilerin ender görülen tiplerinden biridir. Kafatası, çene ve yüz bulgularının yanı sıra görülebilen beyin ve iskelet sistemi bulguları önemlidir.
Abruzzo–Erickson sendromu, sağırlık, dışa çıkık kulaklar, kolobom, yarık damak veya damak rugozitesi, radyal sinostoz ve kısa boy ile karakterize son derece nadir görülen bir hastalıktır. İlk olarak 1977 yılında Abruzzo ve Erickson tarafından, iki erkek kardeş, anneleri ve anne amcalarından oluşan bir ailede değişken şekilde ifade edilen bir CHARGE benzeri sendrom olarak tanımlanmıştır. Bu ailenin üyeleri, CHARGE belirtilerinin birçoğunu sergilemiş, ancak koanal atrezi görülmemiş ve erkek kardeşlerde tipik genital gelişim yaşanmıştır. Bu bozukluğun yakın zamanda keşfedilmesi nedeniyle etiyolojisi tam olarak bilinmemektedir, ancak X-kromozomundaki TBX22 genindeki mutasyonlardan kaynaklandığı düşünülmektedir. Hastalık, X'e bağlı resesif bir şekilde kalıtılır. Şu anda bilinen bir tedavisi yoktur, ancak belirtileri tedavi edilebilir.