Biyofizik, biyolojik olayları incelemek için fizikte geleneksel olarak kullanılan yaklaşım ve yöntemleri uygulayan disiplinler arası bir bilimdir. Biyofizik, moleküler seviyeden organizma ve popülasyon seviyesine kadar tüm biyolojik organizasyon ölçeklerini kapsar. Biyofiziksel araştırmalar biyokimya, moleküler biyoloji, fizikokimya, fizyoloji, nanoteknoloji, biyomühendislik, hesaplamalı biyoloji, biyomekanik, gelişim biyolojisi ve sistem biyolojisi ile önemli ölçüde örtüşmektedir.

Gerhard Heinrich Friedrich Otto Julius Herzberg, Alman asıllı Kanadalı fizikçi ve fiziksel kimyager. 1971 yılında Nobel Kimya Ödülü'nü; "moleküllerin elektron yapısı ve geometri bilgisi ile ilgili katkılarından dolayı, özellikle serbest radikaller alanındaki katkılarından dolayı" kazanmıştır. Herzberg'in ana çalışmaları atomik ve moleküler spektroskopi yöntemleri üzerineydi. Bu teknikleri kullanarak diatomik ve poliatomik moleküllerin yapılarının tanımlanması, başka bir şekilde araştırılması zor olan serbest radikaller ve astronomik yapılarının kimyasal analizi üzerinde çalıştı. Herzberg, 1973 yılından 1980 yılına kadar Kanada, Ottawa'daki Carleton Üniversitesi rektörü olarak görev yaptı.

Moleküler dinamik (MD), atomların ve moleküllerin fiziksel hareketlerini incelemek için bir bilgisayar simülasyon yöntemidir. Atomların ve moleküllerin sabit bir süre boyunca etkileşime girmesine izin verilir ve bu da sistemin dinamik evrimi hakkında bilgi verir. En yaygın versiyonda, atomların ve moleküllerin yörüngeleri, parçacıklar ve bunların potansiyel enerjileri arasındaki kuvvetlerin çoğu zaman atomlararası potansiyeller veya moleküler mekanik kuvvet alanları kullanılarak hesaplandığı, etkileşen parçacıkların bir sistemi için Newton'un hareket denklemlerinin sayısal olarak çözülmesiyle belirlenir. Metot ilk olarak 1950'lerin sonunda teorik fizik alanında geliştirildi, ancak günümüzde çoğunlukla kimyasal fizik, malzeme bilimi ve biyomoleküllerin modellenmesinde uygulanmaktadır.
Bir ajan tabanlı modelleme, sistem üzerindeki etkilerini bir bütün olarak değerlendirmek amacıyla özerk temsilcilerin eylemlerini ve etkileşimlerini taklit etmek için bir hesaplama modelleri sınıfından biridir. Oyun teorisi, kompleks sistemler, ortaya çıkma, hesaplama sosyolojisi, çok etmenli sistemler ve evrimsel programlama unsurlarını birleştirir. Monte Carlo yöntemleri rassallığı tanıtmak için kullanılır. Özellikle ekoloji içerisinde, ABM'lere bireysel tabanlı modeller (IBM) adı da verilir ve IBM'lerdeki bireyler ABM'ler içindeki tamamen özerk ajanlardan daha basit olabilir. Bireysel tabanlı modeller, ajan tabanlı modeller ve çok ajanlı sistemler hakkındaki son literatürün gözden geçirilmesi, ABM'lerin biyoloji, ekoloji ve sosyal bilim de dahil olmak üzere bilgisayarla ilgisiz bilimsel alanlarda kullanıldığını göstermektedir. Ajan tabanlı modelleme, çok etmenli sistemler veya çoklu etmen simülasyonu kavramından farklıdır; ABM' nin amacı, genellikle doğal sistemlerde basit kurallara uyan temsilcilerin ortak davranışlarına açıklayıcı bir bakış açısı bulmaktır.
Hesaplamalı kimya, kimya problemlerini çözmeye yardımcı olmak için bilgisayar simülasyonunu kullanan bir kimya dalıdır. Moleküllerin, katıların yapı ve özelliklerini hesaplamak için verimli bilgisayar programlarına dahil edilmiş teorik kimya yöntemlerini kullanır. Bu yöntemlerin kullanılmasının nedeni, hidrojen moleküler iyonu ile ilgili nispeten yeni sonuçlar dışında, kuantum çok-gövdeli(many-body) problemlerin analitik olarak çözülemez oluşudur. Hesaplama sonuçları normal olarak kimyasal deneylerle elde edilen bilgileri tamamlarken, bazı durumlarda gözlemlenmeyen kimyasal olayları da tahmin edebilmektedir. Yeni ilaç ve materyallerin tasarımında yaygın olarak kullanılmaktadır.

Sistemleri modelleme veya sistem modelleme iş ve bilgi teknolojileri (IT) gelişiminde sistemleri inşa etmek ve kavramsallaştırmak için modellerin kullanımının disiplinlerarası çalışmasıdır.
Moleküler evrim, nesiller boyu aktarılacak şekilde, DNA, RNA ve protein gibi hücresel moleküllerin diziliminin değiştirilmesi işlemidir ya da bununla ilgilenen bilim dalıdır. Moleküler evrimin alanı, bu değişimlerdeki kalıpları açıklamak için evrimsel biyoloji ve popülasyon genetiği ilkelerini kullanır. Moleküler evrim başlıca, nükleotid değişimlerinin oranları ve etkilerini, nötr evrimi, doğal seçilimi, yeni genlerin kökenlerini, karmaşık özelliklerin genetik yapısını, türleşmenin genetik temelini, gelişim evrimini ve evrimin genomik ve fenotipik değişikliklere neden olan etkilerini inceler.
Kuantum kimyası bilgisayar programları, kuantum kimyası metodlarını uygulamak için bilgisayarlı kimyada kullanılır. Çoğu program, Hartree-Fock (HF) ve bazı post Hartree-Fock yöntemlerini içerir ve ayrıca yoğunluk fonksiyonları teorisi (DFT), moleküler mekanik veya yarı-ampirik kuantum kimyası metotlarını da içerebilirler. Bahsi geçen programlar arasında açık kaynaklı ve ticari yazılımlar bulunur. Bunların çoğu büyüktür, çoğu zaman birkaç ayrı program içerir ve uzun yıllar boyunca geliştirilmiştir.

Spartan, Wavefunction'ın moleküler modelleme ve bilgisayarlı kimya uygulamasıdır. Moleküler mekanik, yarı-ampirik yöntemler, ab initio modeller, yoğunluklu fonksiyonel modeller, post Hartree-Fock modeller, G3 (MP2) ve T1 içeren termokimyasal tarifler için kodlar içerir.
Katı hâl fiziğindeki hesaplamalı kimya yöntemleri, moleküller için uyguladıkları aynı yaklaşımı katı hâl fiziğinde de takip eder, ancak bu durumda iki farklılık gösterirler. Birincisi, katının translasyon simetrisinin kullanılması şarttır ve ikincisi, moleküler atom merkezli temel fonksiyonlara bir alternatif olarak düzlem dalgalar gibi tamamen dağılmış taban fonksiyonlarını kullanmak mümkündür. Bir kristalin elektronik yapısı genel olarak Brillouin kuşağındaki her nokta için elektron orbitallerinin enerjilerini tanımlayan bir bant yapısı ile tanımlanmaktadır. Ab initio ve yarı-ampirik hesaplamalar yörünge enerjilerini verir, bu nedenle bant yapı hesaplamalarına uygulanabilirler. Bir molekül için enerjiyi hesaplamak zaman alıcı olduğu için, Brillouin bölgesindeki tüm noktaların listesi için hesap yapmak daha da zaman alıcıdır.

Moleküler modelleme alanında, yanaştırma, bir molekülün ikinci bir moleküle kararlı bir kompleks oluşturmak için birbirine bağlandığında bağlandığında tercih ettiği oryantasyonu tahmin eden bir yöntemdir. Tercih edilen oryantasyon bilgisi iki molekül arasındaki birleşme veya bağlanma afinitesini tahmin etmek için kullanılabilir.

CP2K, katı hal, sıvı, moleküler ve biyolojik sistemlerin atomistik simülasyonlarını gerçekleştirmek için Fortran 2003'te yazılan serbestçe kullanılabilen (GPL) bir programdır.

Moleküler mekanik moleküler sistemleri modellemek için klasik mekaniği kullanır. Born-Oppenheimer yaklaşımının geçerli olduğu varsayılır ve tüm sistemlerin potansiyel enerjisi, kuvvet alanları kullanılarak nükleer koordinatların bir fonksiyonu olarak hesaplanır. Moleküler mekanik, boyutu birkaç atom büyüklüğünde olan sistemlerden tutun da milyonlarca atomdan oluşan büyük sistemlere kadar uygulanabilir.

Atomlar arası potansiyeller, uzayda belirli pozisyonlara sahip atomlardan oluşan bir atom sisteminin potansiyel enerjisini hesaplamak amaçlı kullanılan matematiksel fonksiyonlardır. Atomlar-arası potansiyeller, kimya, moleküler fizik ve malzeme fiziğindeki moleküler mekanik ve moleküler dinamik simülasyonlarının fiziksel temeli olarak çokça kullanılırlar. Bazen kohezyon, termal genleşme ve malzemelerin elastik özellikleri gibi etkilerle bağlantılı olarak kullanılmaktadırlar.
Bu liste, nükleik asit simülasyonları için kullanılan bilgisayar programlarının bir listesidir.

Hesaplamalı biyoloji veya bilişimsel biyoloji, biyolojik, ekolojik, davranışsal ve sosyal sistemlerin araştırılmasında veri analitik ve teorik yöntemlerin, matematiksel modelleme ve hesaplamalı simülasyon tekniklerinin geliştirilmesini ve uygulanmasını içerir. Disiplinlerarası bir alandır ve de farklı disiplinleri kucaklar. Temelinde biyoloji, uygulamalı matematik, istatistik, biyokimya, kimya, biyofizik, moleküler biyoloji, genetik, genomik, bilgisayar bilimi ve evrim yer almaktadır.

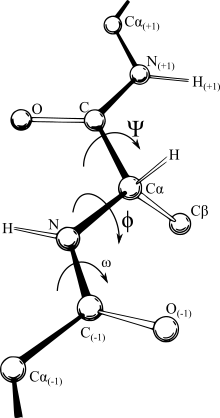
Yapısal biyoloji, biyolojinin özellikle amino asitlerden yapılmış olan proteinler, nükleotitlerden yapılmış RNA ve DNA gibi nükleik asitler ve lipitlerden oluşmuş membranlar olmak üzere biyolojik makromoleküllerin yapılarını ve uzamsal dizilişlerini inceleyen bir dalıdır. Yapısal biyoloji asıl olarak biyofizik yöntemleri ile makromoleküllerin atom düzeyinde üç boyutlu yapılarının belirlenmesi, yapısal değişikliklerinin temel prensipleri, moleküler hareketlerin analizi ve bu yapıların dinamiği ile ilgilenir. Makromoleküller hücrelerin hemen hemen tüm işlevlerini yerine getirir ve bunu da yapabilmek için belirli üç boyutlu şekillere girerler. Moleküllerin "üçüncül yapı"sı olarak adlandırılan bu yapılar her molekülün temel bileşimi ya da "birincil yapı"ları ile karmaşık bir şekilde bağlantılıdır.

Matematiksel ve teorik biyoloji, biyolojinin bilimsel teorileri kanıtlamak için gerekli deneyleri yapmakla uğraşan deneysel biyoloji dalının aksine biyolojik sistemlerin yapılarının, gelişimlerinin ve davranışlarının altında yatan ilkeleri araştırmak için yaşayan organizmaların teorik analizlerini, matematiksel modellerini ve soyutlamalarını kullanan bir dalıdır. Bu alan aynı zamanda matematiksel yanını vurgulamak için matematiksel biyoloji ya da biyomatematik ya da biyolojik yanını vurgulamak için ise teorik biyoloji olarak da adlandırılır. Teorik biyolojinin odak noktası daha çok biyolojinin teorik ilkelerinin geliştirilmesi iken matematiksel biyoloji biyolojik sistemlerin incelenmesinde matematiği kullanır ama her iki terim de bazen birbirinin yerine kullanılabilmektedir.

Kimyasal proses modelleme kimya mühendisliği tasarımında kullanılan bir bilgisayar destekli modelleme tekniğidir. Bu teknikte kullanım amacına yönelik hazırlanmış yazılımlar kullanılarak istenilen proses birbirine bağlı üniteler hâlinde tasarlanır ve yazılım yardımıyla simüle edilerek sistemin yatışkın hâl veya dinamik davranışı tahmin edilebilir. Sistemdeki üniteler ve bağlantılar bir proses akış şeması şeklinde gösterilir. Simülasyonlar bir tankta iki maddeyi karıştırmak kadar basit olabileceği gibi, bir biyodizel tesisinin, petrol rafinerisinin, alüminyum oksit rafinerisinin, doğal gaz işleme tesisinin veya bir biyoetanol saflaştırma ünitesinin tasarım ve kontrolü kadar karmaşık olabilir.
Bir molekül düzenleyici, kimyasal yapıların gösterimlerini oluşturmak ve değiştirmek için kullanılan bir bilgisayar programıdır.