Moleküler mekanik

Moleküler mekanik moleküler sistemleri modellemek için klasik mekaniği kullanır. Born-Oppenheimer yaklaşımının geçerli olduğu varsayılır ve tüm sistemlerin potansiyel enerjisi, kuvvet alanları kullanılarak nükleer koordinatların bir fonksiyonu olarak hesaplanır. Moleküler mekanik, boyutu birkaç atom büyüklüğünde olan sistemlerden tutun da milyonlarca atomdan oluşan büyük sistemlere kadar uygulanabilir.
Tüm atomistik moleküler mekanik yöntemleri aşağıdaki özelliklere sahiptir:
- Her atom bir parçacık olarak simüle edilir
- Her parçacığa bir yarıçap (tipik olarak van der Waals yarıçapı), polarizasyon ve sabit bir net yük (genellikle kuantum hesaplamalarından ve/veya deneyden türetilir) atanır
- Bağlı etkileşimler, deneysel veya hesaplanmış bağ uzunluğuna eşit bir denge mesafesine sahip yaylar olarak ele alınır.
Fonksiyonel formu

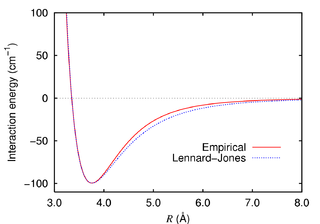
Kimyadaki atomlar arası potansiyel fonksiyon veya kuvvet alanı olarak adlandırılan aşağıdaki fonksiyonel soyutlama, moleküler sistemin potansiyel enerjisini (E) belirli bir konformasyonda bireysel enerji terimlerinin toplamı olarak hesaplar.

kovalent ve kovalent olmayan bileşenleri aşağıdaki özetlerle verilir:


Potansiyel fonksiyonun veya kuvvet alanının tam fonksiyonel formu, kullanılan özel simülasyon programına bağlıdır.
Uygulama alanları
Moleküler mekaniğin ana kullanımı moleküler dinamik alanındadır. Moleküler dinamik, her bir parçacığa etkiyen kuvvetleri hesaplamak için kuvvet alanını ve parçacıkların dinamiklerini modellemek ve yörüngeleri tahmin etmek için uygun bir entegratörü kullanır. Yeterli örnekleme göz önüne alındığında ve ergodik hipoteze tabi olarak moleküler dinamik yörüngeler, bir sistemin termodinamik parametreleri veya reaksiyon hızları ve mekanizmaları gibi kinetik özelliklerini tahmin etmek için kullanılabilir.
Moleküler mekaniğin bir başka uygulaması enerji minimizasyonu olup, kuvvet alanı bir optimizasyon kriteri olarak kullanılır. Bu yöntem, lokal enerji minimumunun moleküler yapısını bulmak için uygun bir algoritma (örneğin en dik iniş) kullanır. Bu minimumlar, molekülün (seçilen kuvvet alanında) kararlı konformerlerine karşılık gelir ve moleküler hareket, bu kararlı konformerler çevresindeki titreşimler ve aralarındaki dönüşümler olarak modellenebilir. Bu nedenle, global enerji minimumunu bulmak için küresel enerji optimizasyonuyla birlikte yerel enerji minimizasyon yöntemlerini bulmak (ve diğer düşük enerji durumlarını) yaygındır. Sonlu sıcaklıkta, molekül zamanının çoğunu düşük enerji seviyelerinde (minimumlarda) geçirir. Bundan dolayı, bu molekülün minimumlardaki hali molekülerin özelliklerinin belirlenmesinde baskındır. Global optimizasyon, benzetilmiş tavlama, Metropolis algoritması ve diğer Monte Carlo yöntemleri kullanılarak veya farklı deterministik ayrık veya sürekli optimizasyon yöntemleri kullanılarak gerçekleştirilebilir. Kuvvet alanı, serbest enerjinin sadece entalpik bileşenini temsil ederken (ve enerji minimizasyonu sırasında sadece bu bileşen dahil edilir), normal mod analizi gibi ek yöntemler kullanılarak entropik bileşeni dahil etmek de mümkündür.
Bu zamana kadar moleküler mekanik potansiyel enerji fonksiyonları bağlanma sabitleri, protein katlama kinetiği, proton dengesi, aktif bölge koordinatlarını hesaplamak ve bağlanma yerlerini tasarlamak için kullanılmıştır.
Yazılım paketleri
Bu sınırlı bir listedir; daha birçok paket mevcuttur.
Ayrıca bakınız
- Moleküler grafikler
- Moleküler dinamik
- Molekül editörü
- Kuvvet alanı (kimya)
- Kuvvet alanı uygulamalarının karşılaştırılması
- Moleküler tasarım yazılımı
- GPU'da moleküler modelleme
- Moleküler mekanik modelleme için yazılımların karşılaştırılması
- Monte Carlo moleküler modellemesi için yazılım listesi
Notlar
- ^ "ACEMD - GPU MD". 21 Kasım 2008 tarihinde kaynağından arşivlendi. Erişim tarihi: 21 Kasım 2008.
- ^ "Ascalaph". 24 Şubat 2010 tarihinde kaynağından arşivlendi. Erişim tarihi: 6 Ocak 2020.
- ^ "COSMOS". 30 Aralık 2008 tarihinde kaynağından arşivlendi. Erişim tarihi: 6 Ocak 2020.
- ^ "CytoSolve". 11 Ekim 2010 tarihinde kaynağından arşivlendi. Erişim tarihi: 6 Ocak 2020.
- ^ "StruMM3D (STR3DI32)". 1 Aralık 2009 tarihinde kaynağından arşivlendi. Erişim tarihi: 6 Ocak 2020.
- ^ "Zodiac". 16 Aralık 2009 tarihinde kaynağından arşivlendi. Erişim tarihi: 6 Ocak 2020.
Kaynakça
- Allinger, N.L.; Burkert, Ulrich, (Ed.) (1982). Molecular Mechanics. An American Chemical Society Publication. ISBN 978-0-8412-0885-8.
- Box VG (March 1997). "The Molecular Mechanics of Quantized Valence Bonds". J Mol Model. 3 (3): 124–41. doi:10.1007/s008940050026.
- Box VG (12 November 1998). "The anomeric effect of monosaccharides and their derivatives. Insights from the new QVBMM molecular mechanics force field". Heterocycles. 48 (11): 2389–417. doi:10.3987/REV-98-504.
- Box VG (2004). "Stereo-electronic effects in polynucleotides and their double helices". J Mol Struct. 689 (1–2): 33–41. Bibcode:2004JMoSt.689...33B. doi:10.1016/j.molstruc.2003.10.019.
- Becker, O.M. (2001). Computational biochemistry and biophysics. New York, N.Y.: Marcel Dekker. ISBN 978-0-8247-0455-1.
- Mackerell AD (October 2004). "Empirical force fields for biological macromolecules: overview and issues". J Comput Chem. 25 (13): 1584–604. doi:10.1002/jcc.20082. PMID 15264253.
- Schlick, T. (2002). Molecular modeling and simulation: an interdisciplinary guide. Berlin: Springer. ISBN 978-0-387-95404-2.
- K. I. Ramachandran; Gopakumar Deepa; Krishnan Namboori (13 Haziran 2008). Computational Chemistry and Molecular Modeling (İngilizce). Springer Science & Business Media. ISBN 978-3-540-77302-3. 9 Ağustos 2019 tarihinde kaynağından arşivlendi. Erişim tarihi: 6 Ocak 2020.
Dış bağlantılar
- Molecular dynamics simulation methods revised
- Molecular mechanics - it is simple28 Aralık 2019 tarihinde Wayback Machine sitesinde arşivlendi.