Miks bağ dokusu hastalığı
| Karışık bağ dokusu hastalığı | |
|---|---|
| Diğer adlar | Sharp sendromu[1] |
| Uzmanlık | İMmünoloji, Romatoloji |
| Ayırıcı tanı | CPT2. |
Genellikle MCTD olarak kısaltılan karışık bağ dokusu hastalığı, şimdi anti-U1 ribonükleoprotein (RNP) olarak adlandırılan belirli bir otoantikorun yüksek kan seviyelerinin varlığı ile birlikte sistemik lupus eritematozus (SLE) semptomlarının bir karışımı ile karakterize edilen bir otoimmün hastalıktır. skleroderma ve polimiyozit.[2] "Karışık" hastalığın arkasındaki fikir, bu spesifik otoantikorun, sistemik lupus eritematozus, polimiyozit, skleroderma, vb. gibi diğer otoimmün hastalıklarda da mevcut olmasıdır. MCTD, 1972'de Sharp ve diğerleri tarafından bireysel bir hastalık olarak karakterize edildi[3][4] ve terim Leroy tarafından tanıtıldı[5] 1980 yılında.[6]
Bazen farklılaşmamış bağ dokusu hastalığı ile aynı olduğu söylenir[1] ancak diğer uzmanlar bu fikri özellikle reddediyor[7] çünkü farklılaşmamış bağ dokusu hastalığı mutlaka U1-RNP'ye yönelik serum antikorları ile ilişkili değildir ve MCTD daha net bir şekilde tanımlanmış bir dizi belirti/semptom ile ilişkilidir.[7]
Belirti ve bulgular
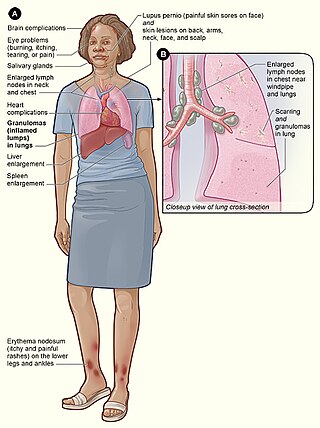
MCTD skleroderma, polimiyozit, sistemik lupus eritematozus ve romatoid artritin özelliklerini birleştirir[8] (bazı kaynaklarda miyozit, dermatomiyozit ve inklüzyon cisimciği miyoziti)[9] eklenir ve bu nedenle bir örtüşme sendromu olarak kabul edilir.
MCTD'nin ilk klinik belirtileri genellikle spesifik değildir. Genel halsizlik, artralji, miyalji ve ateşten oluşabilirler. Bu hastalıktan şüphelenmek için spesifik işaretler, Raynaud fenomeni ile ilişkili, özellikle anti-RNP olan pozitif antinükleer antikorların (ANA) varlığıdır.[10] Hemen hemen her organ MCTD'den etkilenebilir.[11] Raynaud fenomeni hastalarda görülen en yaygın başvuru semptomudur, artralji ve şişmiş eller sırasıyla ikinci ve üçüncü en sık görülen semptomdur.[12] MCTD kriterlerini tam karşılayan hastalarda, azalan sırada takip eden Raynaud, şişmiş eller, lökopeni/lenfopeni ve mide ekşimesi ile artrit en sık görülen semptomdur. 2016 epidemiyolojik popülasyona dayalı bir çalışma, hastalığın ilk belirtilerinden tüm tanı kriterlerinin karşılanmasına kadar geçen ortalama sürenin 3,6 yıl olduğunu bulmuştur.[12]
Belirtiler şunları içerir:
- Deri: Raynaud fenomeni evrenseldir ve neredeyse her zaman hastalık seyrinin başlangıcında mevcuttur. Yokluk, teşhisi sorgular. Kılcal değişiklikler sklerodermanınkine benzer. LES ve sklerodermada görülen tiplere benzer başka deri değişiklikleri de görülebilir.[]
- Artrit: Şişmiş parmaklar ve bazen yaygın ödem belirgin belirtilerdir. Artrit genellikle SLE'de gözlenenden daha sık ve şiddetlidir. Yaklaşık %60'ında belirgin bir artrit ve romatoid artritte gözlenenlere benzer deformiteler bulunur. []
- Miyozit: Miyaljiler – kas ağrıları ve ağrıları – yaygındır, ancak hastaların çoğunda saf polimiyozitte olduğu gibi kas zayıflığı, elektromiyografik değişiklikler ve kas enzimlerinde yükselmeler gözlenmez.[]
- Kalp hastalığı: Perikardit, hastaların %10-30'unda gözlenen en yaygın kardiyak bulgudur. İletim anomalilerinin yanı sıra genellikle pulmoner hipertansiyona sekonder miyokard tutulumu da görülebilir.
- Akciğer tutulumu: Hastaların %75'inde görülür. Plevral efüzyon, pulmoner hipertansiyon, interstisyel akciğer hastalığı, tromboembolik hastalık ve diğerleri olarak ortaya çıkabilir.[]
- Böbrek hastalığı: Şiddetli böbrek hastalığının olmaması, MCTD'nin bir belirtecidir. Bazı durumlarda membranöz nefropati görülebilir.[]
- Gastrointestinal hastalık: En yaygın değişiklik, sklerodermada gözlenene benzer özofagus motilitesindeki değişikliktir.
- Merkezi sinir sistemi tutulumu (CNS): Bu hastalığın orijinal tanımı, CNS'de değişiklik olmadığını vurguladı; bununla birlikte, MCTD'li hastalarda trigeminal nöropatiler (kraniyal sinir V), sensörinöral işitme kaybı ve baş ağrıları gözlenmiştir.[]
- hematolojik anomaliler: Hafif anemi ve hipergamaglobulinemi varlığı yaygındır, SLE'de gözlenenler gibi diğer hematolojik anomaliler de gözlenebilir.[]
- Laboratuvar değer değişiklikleri: Hastaların %50-70'inde romatoid faktör pozitiftir ve hastaların %50'sinde anti-sitrüline protein antikoru saptanır. MCTD'li hastalarda evrensel serolojik bulgular, anti-nRNP özgüllüğü olan anti-nükleer antikorun, özellikle protein 68 kD'ye karşı antikorların varlığıdır.[]
Genetikler
Genetiğin MCTD geliştirmeye katkısı bilinmiyor.[13] Aile üyelerinin MCTD geliştirdiği bilinmektedir, bu da genetiğin MCTD'de rol oynayabileceğini düşündürür, ancak çoğu vaka bireysel olarak ortaya çıkar.[14] MCTD, komorbid bağ dokusu hastalıkları ile ortaya çıkabileceğinden, genetik bir bağlantı olmalıdır, ancak henüz keşfedilmemiştir. DNA metilasyonu, bu hastalığın henüz bilinmeyen genetik risklerini etkileyebilir, çünkü MCTD'li hastalar, sağlıklı meslektaşlarının aksine DNA metilasyon seviyelerini düşürmüştür.[]
Patofizyoloji
MCTD bir otoimmün bozukluktur. RNP çekirdeğin dışında bulunduğunda, RNP'ye karşı anti-RNP antikorları gelişir. RNP, konumu nedeniyle immünolojik olarak korunur, ancak bir hücre ölürse ve RNP artık çekirdekte bulunmuyorsa ve dolayısıyla korumasızsa, bağışıklık sistemi hücresel taklit nedeniyle antikorlar oluşturarak yanıt verebilir. Vücudun geçmişte RNP'ye benzer yapıya sahip moleküllere veya virüslere maruz kalması durumunda MCTD geliştirme riski artabilir.[15]
Şu anda MCTD'ye katkıda bulunan bilinen hiçbir çevresel faktör veya tetikleyici yoktur.[]HLA-DR4 ile ilişkilendirilmiştir.[16]
Epidemiyoloji
MCTD prevalansı dermatomiyozitten daha yüksek ve SLE'den daha düşüktür. 2011 yılında Norveç'te yapılan bir çalışmada, MCTD'nin prevalansı 100.000 yetişkinde 3.8 idi ve insidans yılda milyonda 2.1 idi.[17]
MCTD, kadınlarda erkeklere göre 3: 1 ila 16: 1 oranında ve 50 yaşından küçük kadınlarda çok daha sık görülür.[18] Genel başlangıç yaşı 15-25 yaş civarındadır.[]
Prognoz
Hastalığın orijinal tanımı, genellikle iyi bir prognoz ve kortikosteroidlerle tedaviye mükemmel bir yanıt ile karakterize edilir; ancak gerçekte morbidite ve mortalitesi yüksek bir grup hasta olduğu açıktır. Yakın tarihli bir çalışmada, ölümlerin ana nedenleri pulmoner hipertansiyon, kardiyovasküler problemler ve enfeksiyonlar olmak üzere 5, 10 ve 15 yıllık sağkalım oranları sırasıyla %98, %96 ve %88 idi.[19] Antikardiyolipin antikorlarının varlığı, hastalık için daha ciddi bir risk faktörü olmasının yanı sıra daha fazla skleroderma ve polimiyozit belirti ve semptomlarının varlığıdır.[20]
MCTD'li hastalarda morbidite oldukça yüksektir. Yorgunluk ve tekrarlayan kas-iskelet şikayetlerine ek olarak, orta-yüksek doz kortikosteroid gerektiren ara sıra ortaya çıkan salgınlar sonucunda hastalarda fibromiyalji semptomu gelişebilir. Steroidler, yan etkileriyle birlikte sıklıkla fibromiyalji semptomlarına neden olur ve bu nedenle tedaviyi zorlaştırır.[20] Karışık bağ dokusu hastalığının prognozu, vakaların üçte birinde sistemik lupus eritematozustan (SLE) daha kötüdür. Prednizon tedavisine rağmen, bu hastalık ilerleyicidir ve birçok durumda, kötü bir sonucu olan yaygın kutanöz sistemik skleroderma (dcSSc) olarak da adlandırılan ilerleyici sistemik skleroza (PSS) dönüşebilir. Bazı durumlarda, hastalık hafiftir ve tedavi olarak yalnızca aspirine ihtiyaç duyabilir ve Anti-U1-RNP antikorlarının tespit edilmediği durumlarda remisyona girebilir, ancak bu nadirdir veya vakaların %30'undadır.[] MCTD'den ölümlerin çoğu, pulmoner arteriyel hipertansiyonun (PAH) neden olduğu kalp yetmezliğinden kaynaklanmaktadır.
Hastalık seyri
MCTD teşhisi konan hastalar, SLE, skleroderma veya romatoid artrit gibi diğer bağ dokusu hastalıkları ile daha tutarlı bir klinik tabloya ilerleyebilir. Bazı çalışmalarda bu hastalar, vakaların %9'unda romatoid artrit, %15'inde SLE ve %21'inde skleroderma gibi diğer hastalıklarla zaman içinde yeniden sınıflandırılır.[21] Bu ilerleme kısmen genetik olarak belirlenir, bu nedenle HLA-DR3 hastalarında SLE ve HLA-DR5 hastalarında skleroderma olasılığı daha yüksektir.[20]
Tedavi
MCTD başlangıçta kortikosteroidlere iyi bir tedavi yanıtı olan bir hastalık olarak tanımlansa da, hastalığın tedavisi, diğer bağ dokusu hastalıklarında diğer belirti ve semptomların nasıl tedavi edildiğine benzer şekilde spesifik belirtilere ve klinik komplikasyonlara dayanır.[22][23]
Standart
Artrit için genellikle metotreksat veya hidroksiklorokin ile birlikte kullanılabilen steroid olmayan antienflamatuarlar veya düşük doz prednizon kullanılır. Temporomandibular eklem artritinin, kondral greftler kullanılarak kondil rekonstrüksiyonu ile başarılı bir şekilde tedavi edildiği gösterilmiştir.[24] Miyozit, menenjit, plörit, perikardit, miyokardit, interstisyel akciğer hastalığı veya hematolojik anormallikler gibi komplikasyonlarda daha yüksek dozlarda kortikosteroidler (0,25 ila 1 mg/kg/gün) kullanılır. Aksine Raynaud fenomeni, akroskleroz veya periferik nöropatiler genellikle kortikosteroidlere dirençlidir. Siklofosfamid, interstisyel akciğer hastalığında ve nihai olarak ciddi renal tutulumda faydalıdır. Kortikosteroidlere dirençli miyozit veya trombositopeni vakalarında intravenöz immünoglobulinler faydalı olabilir. Raynaud için genel önlemler (tütünün kesilmesi, soğuğa karşı koruma gibi), kalsiyum antagonistleri, endovenöz prostaglandinler veya endotelin-2 antagonistleri faydalı olabilir. Gastroözofageal reflü olan hastalarda, bu skleroderma problemlerinin olağan tedavisi için protokol izlenerek proton pompa inhibitörleri ve H2 reseptör antagonistleri kullanılabilir.[22][23]
Pulmoner hipertansiyon önde gelen ölüm nedeni olduğundan, rutin ekokardiyografi ile erken teşhisi ve endotelin-1 antagonistleri (bosentan), fosfodiesteraz 5 inhibitörleri (sildenafil) veya endovenöz prostasiklinler (epoprostenol) ile tedavinin hızlı bir şekilde başlatılması, morbidite ve mortaliteyi önemli ölçüde iyileştirmeyi başarır.[22][23]
Soruşturma
MCTD için uygun tedavi seçenekleriyle ilgili daha fazla araştırma devam etmektedir. Çeşitli romatoid hastalıkların tedavisi şu anda araştırma aşamasındadır ve benzer belirti ve semptomlarla başvuran hastalarda kullanılma potansiyeline sahiptir. Hastalığın patofizyolojisinin ve ilerlemesinin daha iyi anlaşılması, daha iyi hedeflenmiş tedavi seçenekleri sağlayacaktır.[25]
Teşhis
Ayırt edici laboratuvar özellikleri, pozitif, benekli bir anti-nükleer antikor ve bir anti-U1-RNP antikorudur.[26][27]
MCTD'nin Sharp tarafından 1972 yılındaki orijinal tanımından sonra, MCTD'nin ayrı bir bağ dokusu hastalığı olup olmadığı konusunda bazı tartışmalar vardı; ancak, kırk yıl ve 2000'den fazla yayından sonra, MCTD'nin ayırt edici bir klinik antite olarak kabul edilmesi gerektiği konusunda bir fikir birliği var gibi görünüyor ve bu nedenle romatologların çoğunluğu tarafından bu şekilde kabul ediliyor,[28] hastalık seyrinde başka bir bağ dokusu hastalığına doğru gelişebilecek bir hasta alt grubu olsa da.
Hemen hemen her organ MCTD'den etkilenebilse de, hastalığın MCTD olduğundan şüphelenmeyi diğer bağ dokusu hastalıklarına göre daha olası kılan çeşitli klinik belirtiler vardır:[29]
- Raynaud fenomeni.
- Ödemli eller ve şişmiş parmaklar.
- Artrit, SLE'den daha şiddetli.
- Pulmoner hipertansiyon (pulmoner fibroz olması gerekmez) MCTD'yi SLE ve sklerodermadan ayırır.
- Yüksek seviyelerde anti-RNP antikorları, özellikle protein 68 kD'ye karşı antikorlar.
- Şiddetli böbrek veya CNS hastalığının olmaması.
Hastalığın tanısını standardize etmek için çeşitli kriterler tanımlanmıştır, en çok kullanılanlardan bazıları Alarcón-Segovia'nınkilerdir, ancak evrensel olarak kabul edilmiş kriterler yoktur.[30][31][32] Çok çeşitli belirti ve semptomlar nedeniyle, tanı için en uygun kriterlerin ne olduğu konusunda bir fikir birliği yoktur. Düşük bir anti-RNP antikor titresi ile bile, bir hasta MTD semptomları gösterebilir. Bunun tersi de mümkündür: yüksek anti-RNP antikor titresi olan bir hasta hiçbir semptom göstermeyebilir.[33] Genel olarak kriterler, yüksek titrelerde anti-RNP antikorlarının varlığını, hastalığın bazı karakteristik belirtilerinin varlığını – Raynaud veya şişmiş eller/parmaklar – ve en az iki diğer bağ dokusu hastalığının bazı klinik belirtilerinin – SLE varlığını gerektirir., skleroderma, polimiyozit.[].
| A. serolojik kriterler: Hemaglütinasyon yoluyla > 1:1600 titrede pozitif Anti-RNP |
| B. Clinical Criteria 1. Ellerin ödemi 2. Sinovit 3. miyozit 4. Raynaud fenomeni 5. akroskleroz |
| MCTD ile mevcut: 3 veya daha fazla klinik kriter ile birlikte Kriter A –biri sinovit veya miyozit olmalıdır– |
SLE, skleroderma ve polimiyozitin daha sıralı klinik belirtilerine göre MCTD tanısını koymak için yeterli belirti ve semptomların ortaya çıkması genellikle birkaç yıl alır, bu nedenle sıklıkla, ilk aşamalarda hastalar için en uygun tanı "farklılaşmamış"tır. bağ dokusu hastalığı".[32]
Hastanın yüksek antinükleer antikor titreleri ile birlikte ödemli elleri ve/veya şişmiş parmakları varsa, yüksek bir anti-U1 RNP antikor titresi, MCTD'ye ilerlemenin iyi bir göstergesidir.[34] Bu spesifik antikorun varlığı, MCTD tanısı için olmazsa olmazdır,[32] izole varlığı, bir hastanın MCTD'si olduğunu veya geliştireceğini garanti etmese de. Baskın otoantikorlar antiDNAn, Sm, Scl70 veya Ro ise, hastanın MCTD'den farklı başka bir bağ hastalığı geliştirmesi muhtemeldir. MCTD'nin klinik belirtileri, U1 RNP kompleksinin protein A' ve 68 kD'sine karşı antikorlarla daha yoğun bir şekilde ilişkili görünmektedir. MCTD'li hastalar HLA-DR4 veya HLA-DR2 ile ilişkili olduğundan, bu arada SLE'li olanlar HLA-DR3 ile ilişkili ve sklerodermalı olanlar HLA-DR5 ile ilişkili olduğundan, MCTD'nin tipik fenotipi de kısmen genetik olarak belirlenmiş gibi görünmektedir.[35]
SLE, skleroderma ve MCTD'de, farklı yüzdelerde anti-U1-snRNP'ye karşı antikorlar bulunur. Bu antikorlar çoğu MCTD hastasında bulunur, ancak SLE'nin yalnızca %30-35'inde ve skleroderma hastalarının %2-14'ünde görülür, bu nedenle MCTD'yi diğer bağ dokusu bozukluklarından ayırt etmeye yardımcı olabilirler. SNRNP70'in farklı haplotipleri vardır ve bunlar, MCTD'li hastalarda SLE veya skleroderma hastalarına göre farklılıkları nedeniyle, MCTD'nin ayrı bir hastalık olduğu iddiasını doğrulamaya yardımcı olur. T-G-CT-G haplotipi MCTD hastalarında daha sık görülürken, T-G-C-G haplotipi skleroderma ve SLE'de daha sık görülür.[36]
Ayırıcı tanı CPT2'yi içerir.
Ayrıca bakınız
- Otoimmün rahatsızlığı
- Özbağışıklık
- örtüşme sendromu
- polimiyozit
- Romatizmal eklem iltihabı
- Romatoloji
- skleroderma
- Sistemik lupus eritematoz
Kaynakça
- ^ a b Rapini, Ronald P.; Bolognia, Jean L.; Jorizzo, Joseph L. (2007). Dermatology: 2-Volume Set. St. Louis: Mosby. ISBN 978-1-4160-2999-1.
- ^ Bennett, Robert (2014). "Clinical manifestations of mixed connective tissue disease". www.uptodate.com. 15 Şubat 2011 tarihinde kaynağından arşivlendi. Erişim tarihi: 12 Ekim 2019.
- ^ Ungprasert, Patompong; Crowson, Cynthia S.; Chowdhary, Vaidehi R.; Ernste, Floranne C.; Moder, Kevin G.; Matteson, Eric L. (December 2016). "Epidemiology of Mixed Connective Tissue Disease 1985-2014: A Population Based Study". Arthritis Care & Research. 68 (12): 1843-1848. doi:10.1002/acr.22872. ISSN 2151-464X. PMC 5426802 $2. PMID 26946215.
- ^ Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR (February 1972). "Mixed connective tissue disease--an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA)". The American Journal of Medicine. 52 (2): 148-59. doi:10.1016/0002-9343(72)90064-2. PMID 4621694.
- ^ Tsokos, George C.; Gordon, Caroline; Smolen, Josef S. (2007). Systemic lupus erythematosus: a companion to Rheumatology. Elsevier Health Sciences. ss. 429-. ISBN 978-0-323-04434-9.
- ^ LeRoy EC, Maricq HR, Kahaleh MB (March 1980). "Undifferentiated connective tissue syndromes". Arthritis and Rheumatism. 23 (3): 341-3. doi:10.1002/art.1780230312
 . PMID 7362686.
. PMID 7362686. - ^ a b Hoffman, Robert W. (1 Haziran 2009). "Mixed Connective Disease". Stone, John (Ed.). Pearls & Myths in Rheumatology. Springer. ss. 169-172. ISBN 978-1-84800-933-2. Erişim tarihi: 26 Haziran 2010.
- ^ "Mixed Connective Tissue Disease, MCTD". The Free Dictionary by Farlex. 13 Eylül 2011 tarihinde kaynağından arşivlendi.
- ^ Nevares, Alana M.; Larner, Robert. "Mixed Connective Tissue Disease (MCTD): Autoimmune Disorders of Connective Tissue". Merck Manual Home Health Handbook. 13 Ekim 2011 tarihinde kaynağından arşivlendi.
- ^ Kaynak hatası: Geçersiz
<ref>etiketi;pmid46216942isimli refler için metin sağlanmadı (Bkz: ) - ^ Kaynak hatası: Geçersiz
<ref>etiketi;:22isimli refler için metin sağlanmadı (Bkz: ) - ^ a b Kaynak hatası: Geçersiz
<ref>etiketi;:32isimli refler için metin sağlanmadı (Bkz: ) - ^ Kaynak hatası: Geçersiz
<ref>etiketi;:0isimli refler için metin sağlanmadı (Bkz: ) - ^ Yang, Chia-Fu; Chiu, Jih-Yu; Su, Chang-Wei; Chen, Chun-Ming (2019). "Chondral grafts for condylar reconstruction in treatment of temporomandibular joint arthritis in a mixed connective tissue disease patient". The Kaohsiung Journal of Medical Sciences. 35 (12): 787-788. doi:10.1002/kjm2.12128 . ISSN 2410-8650. PMID 31512336.
- ^ "Mixed Connective Tissue Disease (MCTD)". NORD (National Organization for Rare Disorders). 25 Mayıs 2015 tarihinde kaynağından arşivlendi. Erişim tarihi: 12 Ekim 2019.
- ^ Aringer M, Steiner G, Smolen JS (August 2005). "Does mixed connective tissue disease exist? Yes". Rheumatic Disease Clinics of North America. 31 (3): 411-20, v. doi:10.1016/j.rdc.2005.04.007. PMID 16084315.
- ^ Gunnarsson, Ragnar; Molberg, Oyvind; Gilboe, Inge-Margrethe; Gran, Jan Tore; PAHNOR1 Study Group (June 2011). "The prevalence and incidence of mixed connective tissue disease: a national multicentre survey of Norwegian patients". Annals of the Rheumatic Diseases. 70 (6): 1047-1051. doi:10.1136/ard.2010.143792. hdl:10852/36041 . ISSN 1468-2060. PMID 21398332.
- ^ "Mixed connective tissue disease - Symptoms and causes". Mayo Clinic. 22 Aralık 2017 tarihinde kaynağından arşivlendi. Erişim tarihi: 12 Ekim 2019.
- ^ Hajas, Agota; Szodoray, Peter; Nakken, Britt; Gaal, Janos; Zöld, Eva; Laczik, Renata; Demeter, Nora; Nagy, Gabor; Szekanecz, Zoltan; Zeher, Margit; Szegedi, Gyula (July 2013). "Clinical course, prognosis, and causes of death in mixed connective tissue disease". The Journal of Rheumatology. 40 (7): 1134-1142. doi:10.3899/jrheum.121272. ISSN 0315-162X. PMID 23637328.
- ^ a b c Bennett, Robert. "Prognosis and treatment of mixed connective tissue disease". 15 Şubat 2011 tarihinde kaynağından arşivlendi.
- ^ Ruiz Pombo, Mónica; Labrador Horrillo, Moisés; Selva O'Callaghan, Albert (20 Kasım 2004). "[Undifferentiated, overlapping and mixed connective tissue diseases]". Medicina Clinica. 123 (18): 712-717. doi:10.1016/s0025-7753(04)75337-3. ISSN 0025-7753. PMID 15563821.
- ^ a b c Mobasat, A.; Turrión Nieves, A.; Bohorquez Heras, C. (January 2013). "Enfermedad mixta del tejido conectivo. Síndromes de solapamiento". Medicine - Programa de Formación Médica Continuada Acreditado. 11 (32): 1991-1996. doi:10.1016/s0304-5412(13)70567-5. ISSN 0304-5412.
- ^ a b c Kaynak hatası: Geçersiz
<ref>etiketi;:5isimli refler için metin sağlanmadı (Bkz: ) - ^ Yang, Chia‐Fu; Chiu, Jih‐Yu; Su, Chang‐Wei; Chen, Chun‐Ming (11 Eylül 2019). "Chondral grafts for condylar reconstruction in treatment of temporomandibular joint arthritis in a mixed connective tissue disease patient". The Kaohsiung Journal of Medical Sciences. 35 (12): 787-788. doi:10.1002/kjm2.12128 . ISSN 1607-551X. PMID 31512336.
- ^ Kaynak hatası: Geçersiz
<ref>etiketi;:72isimli refler için metin sağlanmadı (Bkz: ) - ^ Venables PJ (2006). "Mixed connective tissue disease". Lupus. 15 (3): 132-7. doi:10.1191/0961203306lu2283rr. PMID 16634365. 28 Ağustos 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 20 Ağustos 2022.
- ^ Sato T, Fujii T, Yokoyama T, Fujita Y, Imura Y, Yukawa N, Kawabata D, Nojima T, Ohmura K, Usui T, Mimori T (December 2010). "Anti-U1 RNP antibodies in cerebrospinal fluid are associated with central neuropsychiatric manifestations in systemic lupus erythematosus and mixed connective tissue disease". Arthritis and Rheumatism. 62 (12): 3730-40. doi:10.1002/art.27700. hdl:2433/142082 . PMID 20722023.
- ^ Cappelli, Susanna; Bellando Randone, Silvia; Martinović, Dušanka; Tamas, Maria-Magdalena; Pasalić, Katarina; Allanore, Yannick; Mosca, Marta; Talarico, Rosaria; Opris, Daniela; Kiss, Csaba G.; Tausche, Anne-Kathrin (February 2012). ""To be or not to be," ten years after: evidence for mixed connective tissue disease as a distinct entity". Seminars in Arthritis and Rheumatism. 41 (4): 589-598. doi:10.1016/j.semarthrit.2011.07.010. ISSN 1532-866X. PMID 21959290.
- ^ Kaynak hatası: Geçersiz
<ref>etiketi;:23isimli refler için metin sağlanmadı (Bkz: ) - ^ Alarcón-Segovia, D.; Cardiel, M. H. (March 1989). "Comparison between 3 diagnostic criteria for mixed connective tissue disease. Study of 593 patients". The Journal of Rheumatology. 16 (3): 328-334. ISSN 0315-162X. PMID 2724251.
- ^ "Sociedad Española de Reumatología :: Criterios diagnósticos". 10 Ağustos 2014. 10 Ağustos 2014 tarihinde kaynağından arşivlendi. Erişim tarihi: 12 Ekim 2019.
- ^ a b c Bennett, Robert. "Definition and diagnosis of mixed connective tissue disease". 7 Şubat 2011 tarihinde kaynağından arşivlendi.
- ^ Alves, Marta (2020). ""Mixed connective tissue disease": a condition in search of an identity". Clinical and Experimental Medicine. 20 (2): 159-166. doi:10.1007/s10238-020-00606-7. PMC 7181542 $2. PMID 32130548.
- ^ Greidinger, Eric L.; Hoffman, Robert W. (August 2005). "Autoantibodies in the pathogenesis of mixed connective tissue disease". Rheumatic Disease Clinics of North America. 31 (3): 437-450, vi. doi:10.1016/j.rdc.2005.04.004. ISSN 0889-857X. PMID 16084317.
- ^ Gendi, N. S.; Welsh, K. I.; Van Venrooij, W. J.; Vancheeswaran, R.; Gilroy, J.; Black, C. M. (February 1995). "HLA type as a predictor of mixed connective tissue disease differentiation. Ten-year clinical and immunogenetic followup of 46 patients". Arthritis and Rheumatism. 38 (2): 259-266. doi:10.1002/art.1780380216. ISSN 0004-3591. PMID 7848317.
- ^ "Table 1: The Single Nucleotide Polymorphisms in cathepsin B protein mined from literature (PMID 16492714)". doi:10.7717/peerj.7425/table-1 .