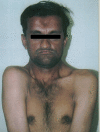
Mikrognati
Mikrognati (micrognathism), altçenenin yetersiz gelişmesi (hipoplazi) niteliğinde bir anomalidir; 1. ve 2. faringeal arklara (brankial yarık) özgü malformasyonların çoğu çene-yüz bölgesindeki mezenkimal dokunun embriyolojik dönemdeki yetersizliğine bağlanmaktadır.[1][2][3][4] Altçene küçüktür ve geridedir; bu olgu, çevredeki kasların hareketlerini sınırlarken bu kaslardan yararlanan dokuların gelişmeleri de kısıtlanır. Kraniyofasiyal anomalileri içeren sendromlarda çok sık görülen bulgulardan biridir.[5][6][7] Sendroma-özgü olmayan olguların varlığı da bilinmektedir; örneğin; altçenesi, kafa ile göğüs kafesi arasında sıkışmış olan bir fetüste çene kemiklerinin gelişimi olması gereken düzeye ulaşamamaktadır.[1][8] Gebelik sorunları, gebelerin alkol kullanması, gestasyonel diabet (gebelik diabeti) gibi çevresel faktörler etkili olabilmektedir (Möbius sendromu).[9] Olguların bir bömümü, gebelerdeki rubella (kızamıkçık) infeksiyonu komplikasyonudur.[10] Yenidoğanların bir bölümünde görülebilen hafif mikrognatiler, altçene gelişiminin tamalanmasıyla silinebilir.[2] Mikrognati'de altçene gövdesi kadar çene eklemini oluşturan yapıların hipoplazisi de önemlidir. Mikrognatilerin bir bölümü kulak sistemi, üstçene ve damak gelişiminin de aksadığı olgularla birlikte görülür.[2][4][5][6][7]
Mikrognati saptanan sendromlar
Total mikrognatiler
Altçenenin tümüyle etkilendiği bir hipoplazinin sonucudurlar. Bu tür mikrognatilerin çoğu sendroma-özgü bir bulgu olarak karşımıza çıkmaktadır; bu sendromların başlıcaları şunlardır: Pierre Robin sequence (sendromu), auriculocondylar sendrom, kraniyofasiyal mikrosomi, Treacher Collins sendromu, Stickler sendromu, Aglossia-adactylia sendromu, Agnati-Otosefali Kompleksi, Carey-Fineman-Ziter sendromu, Glossopalatine ankylosis sendromu, Hallermann-Streiff sendromu, Mandibulofacial dysostosis sendromları, SSASKAS sendromu, Yarık damak sendromu, Williams sendromu, Catel-Manske sendromu, Kedi ağlaması (cri du chat) sendromu, DiGeorge sendromu, Marfan syndrome, Noonan sendromu, Prader-Willi sendromu, Russel-Silver sendromu, Smith-Lemli-Opitz sendromu, Patau sendromu, Edwards sendromu, Wolf-Hirschhorn sendromu.
Mikroretrognati
Retrognati, altçenenin geride olması olgusudur. Hipoplazinin ileri düzeylerde olduğu olgularda altçenenin aşırı biçimde geride olduğu izlenimi alınır; kimi uzmanlar bu tabloyu “mikroretrognati" olarak niteler. Mikroretrognati için örnek sendromlar: Short-rib thoracic dysplasia 13, Verloes-David-Pfeiffer mezomelik displazi, Warkany sendromu, Treacher Collins sendromu.
Retrognati olgularının bir bölümünde ise üstçene iridir (maksiller makrognati) ve altçene geriye itilmiş gibi algılanır. Altçene retrognatisi için örnek sendromlar: 22q11.2 deletion sendromu, Bohring-Opitz sendromu, Diamond-Blackfan anemisi, Hennekam lenfektazi/lenfödem sendromu, Marshall-Smith sendromu, Oral-Facial-Digital sendrom I, Rubinstein-Taybi sendromu, Seckel sendromu, Weaver sendromu, Hutchinson-Gilford progeria sendromu
Ramus hipoplazisi kökenli mikrognatiler
Mikrognatilerin bazıları altçenenin tümünün değil bazı anatomik bölümlerinin hipoplazisinden kökenlidir. Altçenenin, çene eklemini yapmak üzere yukarı dönen parçası olan bölümünün (ramus) hipoplazisi sonucunda oluşan mikrognatilerdir. Örnekler: Hipomandibular fasiyokraniyal disostoz, Ramus hipoplazisi, Hallermann-Streiff sendromu, Auriculocondylar sendrom Andersen-Tawil sendromu.
Ramus ve kondil aplazisi ya da yapısal bozukluğundan kökenli mikrognatiler
Mikrognatilerin bazıları altçenenin tümünün değil bazı anatomik bölümlerinin hipoplazisinden kökenlidir. Altçene ramusunun ve çene ekleminde önemli bir parça olan kondilin oluşamaması ya da biçim bozukluğu sonucu ortaya çıkan mikrognatilerdir. Örnekler: Craniofacial microsomia, Hallermann-Streiff sendromu.
Kondil aplazisinden ya da malformasyonundan kökenli mikrognatiler
Mikrognatilerin bazıları altçenenin tümünün değil bazı anatomik bölümlerinin hipoplazisinden kökenlidir. Altçene ekleminde önemli bir parça olan kondilin oluşamamasının ya da biçim bozukluğunun sonucu yol açtığı mikrognati türüdür. Örnekler: Auriculocondylar sendrom, Fibrodysplasia ossificans progressiva sendromu, Yarık damak sendromu.
Koronoid çıkıntı agenezi ya da hipoplazisinden kökenli mikrognatiler
Mikrognatilerin bazıları altçenenin tümünün değil bazı anatomik bölümlerinin hipoplazisinden kökenlidir. Altçene ramusunun ve çene ekleminin bir parçası olan koronoid çıkıntının oluşamaması ya da biçim bozukluğu sonucu ortaya çıkan mikrognatilerdir. Örnekler: Auriculocondylar sendrom, Melnick-Needles sendromu.
Altçene atrofisi
Mandibuloacral dysplasia sendromu'nda, başlangıçta belirsiz olan mikrognati zamanla önemli bulgulardan biri niteliğini kazanır; buradaki altçene küçülmesi "atrofi" niteliğindedir.
Kaynakça
- ^ a b Singh DJ, Bartlett SP. Congenital mandibular hypoplasia: analysis and classification. Journal of Craniofacial Surgery, 16:291–300, 2005
- ^ a b c Johnson JM, Moonis G, Green GE, et al. Syndromes of the First and Second Branchial Arches, Part 1: Embryology and Characteristic Defects. American Journal of Neuroradiology, 32 (1) 14-19, 2011
- ^ Sadler TW, Langman J. Langman's Medical Embryology. 10th ed. Lippincott-Williams & WilkinsPhiladelphia, 2006
- ^ a b Stevenson RE, Hall JG.(eds). Human Malformations and Related Anomalies. 3rd edition. Oxford University Press, Oxford-New York, 2015
- ^ a b Mooney MP, Siegel MI. Understanding Craniofacial Anomalies: The Etiopathogenesis of Craniosynostoses and Facial Clefting. Wiley-Liss, New York, 2002
- ^ a b Passos-Bueno MR, Ornelas CC, Fanganiello RD. Syndromes of the first and second pharyngeal arches: A review. American Journal of Medical Genetics, 149A:1853–1859, 2009
- ^ a b DeLuke DM, Haug RH. Syndromes of the Head and Neck. Elsevier, Philadelphia, 2014
- ^ Perrone JA. Micrognathia in Newborns. Archives of Otolaryngology, 90(1):85-86, 1969
- ^ Ghosh R, Shetty V, Hegde S, et al. Rare features associated with Mobius syndrome: Report of two cases. Journal of Dental Research, Dental Clinics, Dental Prospects, 11(1):60-65, 2017
- ^ Yazigi A, De Pecoulas AE, Vauloup-Fellous C, et al. Fetal and Natal abnormalities due to congenital rubella syndrome: a review of literature. Journal of Maternal-Fetal & Natal Medicine, 30(3):274-278, 2017