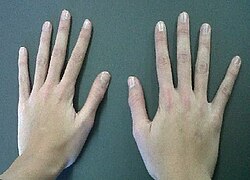
Williams sendromu , 7. kromozomun uzun kolunda 26 genin silinmesiyle ortaya çıkan; ektodermal displazi bulguları da içeren, otosomal dominant geçen kalıtsal bir sendromdur. Genel gelişme geriliği izlenir, hastaların çoğu zayıftır. Kafatasındaki gelişme duraklamasının sonucu olarak oldukça geniş bir alın vardır. Yabancılara kolay güvenme, geç gelişen dil becerileri, kalp rahatsızlığı, geç gelişen koordinasyon-denge becerisi gibi sonuçlar doğuran nörolojik bozukluktur. Algılama (kognitif) sorunları vardır, psikiyatrik bulgularla karşılaşılabilir. Ses telleri felci nedeniyle ses kabadır. Hasta aktiftir ve mutlu bir görünüm ile aşırı dostça davranış sergiler. Uyku sorunları ve zeka geriliği olabilir.
Kraniyoektodermal displazi (Cranioectodermal dysplasia), ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur. 4 fenotipi vardır:
- Cranioectodermal dysplasia 1 (Sensenbrenner sendromu; Levin sendromu; arthrodentoosseous dysplasia)
- Cranioectodermal dysplasia 2
- Cranioectodermal dysplasia 3
- Cranioectodermal dysplasia 4
Ağız-Yüz-Parmak sendromu tip 1 , ektodermal displazi bulguları da içeren, X-kromozomu aracılığıyla dominant (XLD) geçen kalıtsal bir sendromdur. Simpson-Golabi-Behmel sendromu tip 2 ile alelik bağı olduğu belirlenmiştir. Kız çocuklarında görece sıktır. Erkek fetüsler, kalp ve beyin anomalilerinin neden olduğu intauterin ölümler nedeniyle kaybedilirler. Belirgin bir genel gelişme geriliği saptanır.
Gorlin-Cohen sendromu , ektodermal displazi bulgularını da içerebilen bir OPD sendromu tipidir. 2 fenotipi vardır:
- Frontometaphyseal dysplasia 1
- Frontometaphyseal dysplasia 2
Beckwith-Wiedemann sendromu, otosomal dominant yolla aktarılan kalıtsal bir aşırı büyüme sendromudur. 4 fenotipi vardır; Konjenital hemihipertrofi (hemihiperplazi) en önemlisidir. Russell-Silver sendromu'nun temel nedenin 11p15 (ICR1) distal kromozomunun yetersiz metilasyonu (hipometilasyon) ise olduğu gösterilmiş; aynı kromozomun hipermetilasyonunun ise Beckwith-Wiedemann sendromuna yol açabileceği ileri sürülmüştür.

Baraitser-Winter sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; 2 fenotipi vardır. Genel gelişme geriliği izlenir. Kraniyosinostoz nedenli trigonosefali ve mikrosefali saptanır. Boyun kısadır. Hipertelorizm saptanır, kaş çıkıntıları yüksektedir. Göz kapakları iridir ve bilateral ptozis saptanır. Mikroftalmi ile iris ve retina defektleri (koloboma) izlenir. Kulak kepçeleri küçüktür ve aşağıdadır; işitme sorunları vardır. Burun sırtı kalın, ucu büyüktür. Uzun bir filtrum altında ince bir üst dudak bulunur. Alt dudak kalındır. Üst dudak ve damak yarıktır. Altçene geridedir (retrognati). Omurga sisteminde kifoz ve skolyoz türü malformasyonlar saptanır. Başparmak sayısı birden fazladır (polidaktili). Değişik oranlarda konjenital kalp anomalileri ve ürogenital anomaliler görülür. Beyinde corpus callosum anomalileri ile frontal lob anomalisi saptanır; epilepsi ve zeka geriliği bulguları vardır. Genel hipotoninin yanı sıra omuz kaslarında giderek artan güçsüzlük görülür. Deri lenfoması ve lösemi riski yüksektir.
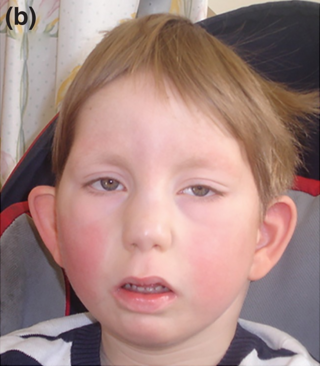
Au-Kline sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; böbrek, kalp ve yüz bulgularıyla öne çıkar.

Cornelia de Lange sendromu , 5 fenotipi olan kalıtsal bir sendromdur. Tüm fenotiplerde benzer bulgular saptanır; farklılık etkilenen genlerdedir.

Emanuel sendromu, t(11;22)(q23;q11.2) ayrımı bozukluğu olarak tanımlanan bir kromozom anomalisi sendromudur.

Miller-Dieker lissensefali sendromu, büyük bölümü otosomal dominant yolla aktarılan kalıtsal mikrosefali türü.

MVA sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. 3 fenotipi vardır; fenotip 1 en sık görülenidir. Anöploidi olarak niterlendirilen kromozom sayısı anomalisinden kökenlidir; kromozom eksikliği (monosomi) ya da fazlalığı (trisomi) vardır. Ancak, bazı hücrelerdeki kromozom sayısının normal olması nedeniyle “mozaisizm” grubu içinde yer alır.

Renpenning sendromu ya da Golabi-Ito-Hall sendromu, X-kromozomu aracılığıyla aktarılan 60'a yakın “mental retardasyon ” sendromlarının fenotiplerinden biridir.
Ritscher-Schinzel sendromu, 2 fenotipi olan kalıtsal bir sendromdur.

Toriello-Carey sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Doğum ölçümleri normaldir, ancak zamanla gelişme geriliği bulguları belirir.

Bohring-Opitz sendromu, otosomal dominant yolla aktarılan, gelişme ve zeka geriliği bulgularının ön planda olduğu, hastaların çoğunun çocukluk yaşlarında kaybedildiği bir sendromdur. C sendromu'nun fenotipi olarak benimsenir.

Meckel-Gruber sendromu, çok sayıda genin etkilenmesinden kaynaklanan, bu nedenle 15 fenotipi olan, otosomal resesif yolla aktarılan, siliyopatiler grubundan, kalıtsal bir sendromdur. Kistik böbrekler, beyin anomalileri ve ensefalosel, polidaktili ve karaciğer anomalileri başlıca bulgulardır. En sık görülen fenotip Meckel sendromu 1, öteki fenotiplerdeki bulguların çok büyük bir bölümünü içerir. Bazı fenotipleri Joubert sendromuyla çakışan nitelikleri içermektedir.

Oculofaciocardiodental sendrom, göz malformasyonları, çene-yüz anomalileri, dolaşım sistemi ve bağışıklık sistemi defektleriyle karakterize, X-kromozomu aracılığıyla dominant (XLD) yolla aktarılan kalıtsal bir sendromdur. Mikroftalmi sendromunun 18 fenotipinden biridir.

Saethre-Chotzen sendromu (acrocephalosyndactylia III), fiziksel gelişme geriliği, kraniyosinostoz nedenli kafatası anomalileri, asimetrik yüz, göz ve parmak malformasyonlarının saptandığı otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Kraniyosinostozun çok sayıda eklemi etkilediği olgularda “kafaiçi basıncı artışı sendromu (KİBAS)” gelişebilir.

Shprintzen-Goldberg sendromu , marfanoid yapı, kraniyosinostoz, kardiyovasküler ve nörolojik anomalilerle karakterize, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Marfanoid yapı, Marfan sendromundaki bulguların büyük bölümünün saptandığı, ancak Marfan sendromunudaki genetik altyapının bulunmadığı olgular için kullanılan nitelemedir; uzun kollar ve bacaklar, örümceksi parmaklar (araknodaktili), eklemlerde aşırı gevşeklik ve mitral kapak prolapsusu Marfan sendromunu andıran başlıca bulgulardır. Robbins Shprintzen-Goldberg sendromu’nun, Marfan sendromu ve Loeys-Dietz sendromu ile oldukça çok büyük benzerlikleri vardır.
Meckel sendromu, birden fazla etkisi olan bir genle ilgili (pleiotropic), otosomal resesif yolla aktarılan, 15 kadar fenotipi bulunan kalıtsal bir sendromdur. Çok sayıda ve karmaşık bulguların varlığı tanıda güçlükler oluşturur. Bazı uzmanlar, Meckel sendromunun 3 temel bulgusu olduğunu savunurlar: (i) Santral sinir sistemi malformasyonları ; (ii) Kistik böbrekler; (iii) Karaciğer patolojileri. Bebeklerin yaşama yetisi çok kısıtlıdır; canlı doğabilenlerin çoğu birkaç hafta içimde yitirilir.