Kraniyosinostoz
Kraniyosinostoz, kraniyosinostozis (craniosynostosis), kraniyofasiyal malformasyonların ve maksillofasiyal (çene ve yüz) sendromların önemli bir bölümünde etkileri görülebilen konjenital bir patolojidir.[1][2] Bu olgudaki temel bulgu kafatası eklemlerinin (kraniyal sutura) erken kapanmasıdır; etkilediği anatomik bölgelerde ortaya çıkan malformasyonlar, hangi suturaların ne düzeyde kapanmış olmasıyla orantılıdır. Malformasyonlar genellikle etkilenen eklemin dikey yönünde belirgindir.[3][4][5][6][7][8]
Sagital kraniyosinostoz (sagittal craniosynostosis), en sık görülen tiptir. Sutura sagittalis’in (iki parietal kemiğin orta çizgi üzerinde eklemleşmesi) erken kapanmasının sonucudur. Bu aksaklık, dar ve uzun bir kafatasının oluşmasıyla sonlanır (dolikosefali). Çoğu kez izole bir olgu olarak ortaya çıkar. Sutura sagittalis’in etkilendiği “scaphocephaly” olgusunda, yüz bölgesi malformasyonların çok hafif olduğu saptanır. Oysa, sutura metopica’nın (frontal kemiklerin buluşma çizgisi) erken kapanmasıyla ortaya çıkan “trigonocephaly” ya da “Kleeblattschadel (yonca yaprağı kafatası)” olgularında, yüz bulguları oldukça güçlüdür; bu gruptaki sendromlara “craniofacial dysostosis sendromları” nitelemesi yapılır.[3][6][9]
Koronal kraniyosinostoz (coronal craniosynostosis), sutura coronalis’in (frontal kemiğin parietal kemiklerle olan eklem çizgisi) erken kapanmasının sonucudur. Sagital kraniyosinostoz’dan sonra ikinci sıklıkta saptanır. İki taraflı (bilateral) olduğunda geniş-yayvan bir kafatası (brakisefali) ile sonlanır. Tek taraflı (unilateral) olgulardaki kafatası deformitesi diyagonaldir (plagiosefali).[3][6][9]
Nedenler
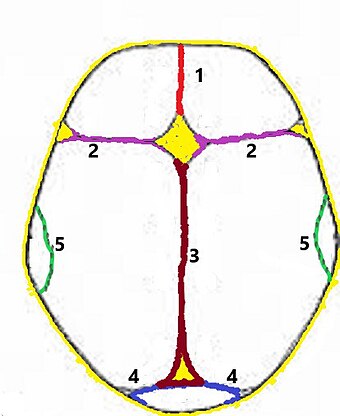
Kraniyosinostozlar, primer ya da sekonder olarak ortaya çıkar. Primer tip kraniyosinostozlardaki prematüre eklem kaynaşmalarında gen mutasyonları ve sendromlar söz konusudur. Sekonder tipler ise, talassemi ve hipertiroidizm gibi bir hastalığa özgü bulgulardan ya da komplikasyonlarından biridir. Spontan olguların bir bölümünde, gebelik sorunlarının (özellikle teratojenler) etkileri saptanmıştır.[8][10][11]
Kraniyosinostoz patogenezi
Kraniyosinostozların oluşumunda 3 görüş öne sürülmektedir:[8][11][12]
(1) Virchow teorisi: Kraniyosinostozlar, kraniyofasiyal malformasyonlarda ortaya çıkan ilk patoloji olarak nitelendirilir. Kraniyosinostoz olgusundan öncelikle kafa tabanı kemikleri etkilenir.
(2) Moss teorisi: Kraniyosinostozlar, kafa tabanı kemiklerindeki malformasyonların sonrasında ortaya çıkan patolojilerdir.
(3) Mezenkimal blastema teorisi: Suturalardaki ve kafa tabanı kemiklerindeki malformasyonların birlikte ortaya çıkmasının sorumlusı “mezenkimal blastemada”ki bir defekttir.
İndüksiyon mekanizması kurallarına göre, kafatası kemikleri beyin dokusunun (özellikle dura mater) gelişmesinden etkilenerek gelişirler. Suturalardan biri erken kapandığında, beyin büyüdükçe, kafatası genişlemesinin normal suturalar yönünde gerçekleştiği izlenir; kraniyofasiyal anomalilerde saptanan kafatası malformasyonlarının çok büyük bölümünün temelinde bu mekanizma etkilidir.
Bulgular
Kraniyosinostozis içeren çoğu sendromda saptanan 5 temel bulgu izlenir:[1][3][4][5][6][8]
- Kraniyosinostozis (coronal-sagittal-lambdoid suturalar)
- Maksilla hipoplazisi (mikrognati)
- Aşırı sığ orbitalar
- Proptozis ve ekzoftalmi
- Hidrosefalus
Kraniyosinostozis olgularında kafa tabanı kemikleri sıkça etkilenir; hidrosefalusla birlikte olduğunda nörolojik bulguların daha da belirginleşir, Arnold–Chiari malformasyonu gelişebilir. Sendroma-özgü olmayan olguların çoğunda önemli klinik bir belirti olmayabilir:[1][3][4][5][6][7][13][14]
Tek taraflı (unilateral) sutura kapanmaları “plagiocephaly” olarak nitelendirilen malformasyona neden olur. Yüz orta bölümünün gelişmesi, kafatası ön bölgesinin gelişmesinden önemli derecede etkilenir; bu nedenle, iki taraflı (bilateral) kraniyosinostozda yüz orta bölüm malformasyonları çok güçlüdür (örneğin; Crouzon sendromu, Apert sendromu).
Kraniyosinostozlar, kromozom anomalilerinde ve maksillofasiyal sendromlarda görülen çene-yüz yarıklarının büyük bölümünün de temel nedenlerindendir; bunlardan en önemlisi Crouzon sendromu’dur. Crouzon sendromunda koronal suturalar ile birlikte sagittal-lambdoidal suturalar da kapanmıştır. Bu malformasyon, kafa tabanının gerektiğinden kısa oluşmasına yol açar. Bu sonuç, dışarıya yüz orta bölümünün hipoplazisi olarak yansır. Yüz orta bölümü çökmüştür; orbitalar sığdır ve gözler fırlaktır (eksorbitizm), hafif bir hipertelorizm ile yalancı bir altçene prognatisi saptanır. Arkaya itilmiş olan yumuşak damak oral ve faringeal boşlukların daralmasına neden olur (obstrüktif uyku apnesi nedeni).
Apert sendromu’nda (acrocephalosyndactyly) ise, kafatası kısa ve kule gibidir; geniş bir alın yapısı izlenir (turribrakisefali); koronal suturaların kapanmasına sagittal ve lambdoidal suturalar da eşlik edebilir. Kaşlar üzerinde transvers bir çıkıntı izlenir. Oksipital kemik düzdür. Hipertelorizm oldukça belirgin; eksorbitizm belli belirsizdir. Yüz orta bölümündeki hipoplazi “psödomandibular prognatizm” algısı yaratır. Çukur damak, dar damak ve yarık damak bulguları olabilir. El ve ayak malformasyonları (sindaktili) Apert sendromunun önemli bir parçasını oluşturur.
Pfeiffer sendromu’ndaki maksillofasiyal bulgular, kraniyosinostozlarda saptanan bulgulardan oluşur. Orta yüz hipoplazisi ve turribrakisefalik yüz yapısı baskındır. Hipertelorizm ve eksorbitizm hafiftir. Parmak anomalileri dikkat çekicidir.
Saethre–Chotzen sendromu, koronal suturalraın sinoztozuyla birlikte akrosefalik bir nitelik taşır. Yüz orta bölüm hipoplazisi yoktur; ancak, yüz yapısı tümüyle asimetriktir. Düşük saç çizgisi, ptozis, yayvan burun ve yüz açılarında düzensizlik saptanır. Parmaklar kısadır, sindaktili vardır.
Carpenter sendromu’nda (acrocephalopolysyndactyly) koronal suturaların kaynaşması asimetrik olduğu için kranyumdaki malformasyon da asimetrik bir kule biçimindedir. Parmak anomalileri arasında polidaktili öne çıkar. Konjenital kalp defektleri olabilir.
Yonca yaprağı kafatası (kleeblattschädel): Kafatası üç lobludur. Bu anomalide temporoparietal, koronal, lambdoidal ve metopik suturaların değişen oranlarda katkıları vardır; en az 3 eklemin erken kapanmasının sonucudur (pansinostoz). Hidrosefali, yüz orta bölüm hipoplazisi ve eksorbitizm saptanır.
Kraniyosinostoz içeren sendromlar
Kraniyosinostoz içeren sendromlara örnekler verilmektedir:
Aşırı Brakisefali bulgusu içerenler
- 6pter-p24 deletion sendromu
- Acromelic frontonasal dysostosis
- Apert sendromu
- Baller-Gerold sendromu
- Bardet-Biedl sendromu
- Cornelia de Lange sendromu
- Down sendromu
- Elsahy-Waters sendromu
- Frontofasiyonazal displazi
- Gorlin-Chaudhry-Moss sendromu
- Hallermann-Streiff sendromu
- Kaufman oculocerebrofacial sendromu
- Roberts sendromu
- Scheuthauer-Marie-Sainton sendromu
- Serebrofasiyotorasik displazi
- Weill-Marshesani sendromu
- Zellweger sendromu
- Zunich nöroektodermal sendromu
Cebocephaly (burun yokluğu+hipotelorizm) bulgusu içeren sendrom
- Psödotrisomi 13 sendromu
Aşırı Dolikosefali bulgusu içeren sendromlar
- 3M sendromu
- Beals-Hecht sendromu
- Bloom sendromu
- BOF sendromu
- Cranioectodermal dysplasia kümesi
- Marfan sendromu
- Prader Willi sendromu
- Proteus sendromu
- Pycnodysostosis
- Short-rib thoracic dysplasia 7
- Short-rib thoracic dysplasia 18
- Smith-Fineman-Myers sendromu
- Sotos sendromu
- TDO sendromu
Hiperostozis (kalın-yoğun kafa kemikleri) bulgusu içeren sendromlar
- Alström sendromu (frontal)
- Craniometaphyseal dysplasia (tümü)
- Gorlin-Cohen sendromu (supraorbital ve frontal)
- Hiperfosfatemik tümöral kalsinozis (tümü)
- McCune-Albright sendromu (tümü)
- Melnick-Needles sendromu (tümü)
- Morgagni-Stewart-Morel sendromu (nebula frontalis)
- Maroteaux-Lamy sendromu (tümü)
- Oculodentodigital sendrom (tümü)
- Psödohipoparatiroidizm (tümü)
Kraniyosinostoz (craniosynostosis) içeren sendrom örnekleri
- 3MC sendromu
- 22q11.2 deletion sendromu
- Antley-Bixler sendromu
- Apert sendromu
- Baller-Gerold sendromu
- Cole-Carpenter sendromu
- Craniofrontonasal sendrom
- Crouzon sendromu
- Gorlin-Chaudhry-Moss sendromu
- Hipomandibular fasiyokraniyal disostoz
- Jackson-Weiss sendromu
- Opitz GBBB sendromu Tip2
- van den Ende-Gupta sendromu
- Zhu-Tokita-Takenouchi-Kim sendromu
- Baraitser-Winter cerebrofrontofacial sendromu (trigonosefali)
- Bohring-Opitz sendromu (trigonosefali)
- C sendromu (trigonosefali)
- Greig sefalopolisindaktili sendromu (trigonosefali)
- Jacobsen sendromu (trigonosefali)
- Potocki-Lupski sendromu (trigonosefali)
- Short-rib thoracic dysplasia 9 (trigonosefali)
- Cranioectodermal dysplasia 3 (plagiosefali; asimetrik kafatası)
- Curry-Jones sendromu (plagiosefali; asimetrik kafatası)
- Münke sendromu (plagiosefali; asimetrik kafatası)
- Opitz-Kaveggia sendromu (plagiosefali; asimetrik kafatası)
- Saethre-Chotzen sendromu (plagiosefali; asimetrik kafatası)
- Beare-Stevenson sendromu (yonca yaprağı kafatası)
- Carpenter sendromu (yonca yaprağı kafatası)
- Cranioectodermal dysplasia 2 (yonca yaprağı kafatası)
- Cumming sendromu (yonca yaprağı kafatası)
- Kleeblattschadel sendromu (yonca yaprağı kafatası)
- Osteoglofonik displazi (yonca yaprağı kafatası)
- Pfeiffer sendromu (yonca yaprağı kafatası)
- Shprintzen-Goldberg sendromu (yonca yaprağı kafatası)
Tedavi
Kraniyosinostoz olgularındaki olumsuzluklar, nörolojik bozukluklara yol açtığı kadar entelektüel nitelikleri de etkiler. Yüz bölgesi kemiklerinin de etkilenmesi obstrüktif uyku apnesi bulgularına neden olur. Bu tür olumsuzlukların önlenmesindeki tek çözüm cerrahi girişimlerdir. Kafatası boyutlarının normal sınırlara getirilmesinin yanı sıra dişhekimliğinin katkıları da gerekebilir.[14][15][15][16][17][18]
Kaynakça
- ^ a b c Kimonis V, Gold JA, Hoffman TL, et al. Genetics of craniosynostosis. Seminars in Pediatric Neurology. 14(3): 150–61, 2007
- ^ Priolo M. Ectodermal dysplasias: An overview and update of clinical and molecular-functional mechanisms. American Journal of Medical Genetics, 149A:2003–2013, 2009
- ^ a b c d e Stricker M, Van der Meulen JC, Raphael B, Mazzola R. Craniofacial Malformations. Churchill Livingstone, Edinburgh, 1990
- ^ a b c Cohen MM Jr, Sven Kreiborg S. Perspectives on craniofacial syndromes, Acta Odontologica Scandinavica, 56(6):315-320, 1998
- ^ a b c Gorlin RJ, Cohen MM Jr, Hennekam RC. Syndromes of the Head and Neck. 4th ed., Oxford University Press, New York, 2001
- ^ a b c d e DeLuke DM, Haug RH. Syndromes of the Head and Neck. Elsevier, Philadelphia, 2014
- ^ a b Stevenson RE, Hall JG (editors). Human Malformations and Related Anomalies. 3rd edition. Oxford University Press, Oxford-New York, 2015
- ^ a b c d Yıldız ME. Kraniosinostozlu olgularda radyolojik değerlendirme. Türk Nöroşirurji Dergisi, 27(3):263-277, 2017
- ^ a b Cohen MM Jr. Malformations of the Craniofacial Region: Evolutionary, Embryonic, Genetic, and Clinical Perspectives. American Journal of Medical Genetics (Seminars in Medical Genetics), 115:245–268, 2002
- ^ Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: Understanding genetic and environmental influences. Nature Reviews Genetics, 12:167-178, 2011
- ^ a b Trainor PA, Andrews BT. Facial dysostoses: Etiology, pathogenesis and management. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 163C(4):283-294, 2013
- ^ Mooney MP, Siegel MI. Understanding Craniofacial Anomalies: The Etiopathogenesis of Craniosynostoses and Facial Clefting. Wiley-Liss, New York, 2002
- ^ Ahmed MK, Ye X, Taub PJ. Review of the genetic basis of jaw malformations. Journal of Pediatric Genetics, 5(4):209-219, 2016
- ^ a b Wang JC, Nagy L, Demke JC. Syndromic Craniosynostosis. Facial Plastic Surgery Clinics of North America, 24(4):531-543, 2016
- ^ a b de Jong T, Bannink N, Bredero-Boelhouwer HH, et al. Long-term functional outcome in 167 patients with syndromic craniosynostosis; defining a syndrome-specific risk profile. Journal of Plastic, Reconstructive & Aesthetic Surgery, 63(10): 1635-41, 2010
- ^ Passos-Bueno MR, Ornelas CC, Fanganiello RD. Syndromes of the first and second pharyngeal arches: A review. American Journal of Medical Genetics, 149A:1853–1859, 2009
- ^ Cohen MM, MacLean RE. Craniosynostosis: Diagnosis, Evaluation, and Management, 2nd edition, Oxford University Press, Oxford, 2000
- ^ Persing JA. MOC-PS(SM) CME article: Management Considerations in the Treatment of Craniosynostosis. Plastic and Reconstructive Surgery, 121(4 Suppl): 1–11, 2008