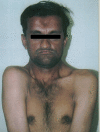
Cleidocranial dysostosis, kemik malformasyonları içeren kalıtsal bir sendromdur; ebeveynlerden birinden otosomal dominant yolla gelir. Genetik incelemeler, kemik gelişimiyle ilgili RUNX2 genindeki bir sorunu göstermektedir. 3 tipi vardır; dişlerin etkilenmesi bunlardan yalnızca birinde saptanır. İskelet sistemi etkilenmesinin en tipik bulgusu köprücük kemiklerinde (klavikula;clavicula) görülür. Köprücük kemikleri ya hiç oluşmamıştır (agenezis) ya da gelişmesi yetersizdir (hipoplazi); bu nedenle, hastalar, omuzlarını orta çizgi üzerinde birleştirebilirler. Kafatasındaki bıngıldaklar ve pelvisteki simfizi uzun süre kapanamaz.
OPD sendromu (otopalatodigital sendrom), X-kromozomu aracılığıyla dominant (XLD) yolla geçen kalıtsal bir sendromdur (Gorlin-Cohen sendromu hariç). Kız çocukları daha hafif etkilenir. Tümü benzer bulgular içeren 5 tipi vardır:
- Otopalatodigital sendrom tip I (OPD tip1)
- Otopalatodigital sendrom tip II (OPD tip2)
- Gorlin-Cohen sendromu (ilgili sayfaya gidiniz)
- Melnick-Needles sendromu (tip 1 ile allelik bağlantısı vardır; ilgili sayfaya gidiniz)
- Terminal osseöz displazi ve pigmentli deri defektleri (çok enderdir, ayrıntı verilmemiştir).
Gorlin-Cohen sendromu , ektodermal displazi bulgularını da içerebilen bir OPD sendromu tipidir. 2 fenotipi vardır:
- Frontometaphyseal dysplasia 1
- Frontometaphyseal dysplasia 2

Marshall sendromu, ektodermal displazi bulguları da içerebilen, otosomal dominant yolla geçen kalıtsal bir sendromdur.
Even-Plus sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Genel gelişme geriliği vardır. Yenidoğanda, deri malformasyonu saptanır. Brakisefalik kafa biçimindeki kafatasında saçlar seyrektir. Boyun kısa, kaş çıkıntıları belirgindir. Kaşlar birbirine birleşmiş olabilir. Kulak deliği yoktur ya da dardır (microtia). Burun hipoplaziktir, ucunda yarık vardır. Yüz orta bölüm hipoplazisi nedeniyle üstçene küçüktür (mikrognati), damak çukuru belirgindir. Üst çenede tek orta kesici (santral) diş vardır, lateral dişler eksiktir (hipodonti). Kalpte konjenital anomaliler saptanır. Anüste darlık olabilir. Üriner sistemde, böbrek hipoplazisi ve infeksiyonlar olabilir. İskelet sisteminde, omurga yarıkları ve koksiks agenezi saptanır; uzun kemiklerde malformasyonlar vardır. Atopik dermatit olabilir. Corpus callosum agenezi, beyinde saptanan en önemli anomalidir.
Diastrofik displazi, otosomal resesif yolla aktarılan kalıtsal bir sendromdur; 2 fenotipi vardır. Genel gelişme geriliği saptanır; yenidoğanda kollar ve bacaklarda belirgin biçimde kısadır (cücelik). Kaşlar bölgesinde (glabella) damar tümörü (hemangioma) görülür. Kulak kepçeleri büyüktür, kistik oluşumlar vardır. İşitme sorunları bulunur. Ağız içi incelemede, yarık damak saptanır. Larinks stenozu nedeniyle ses kabadır. Vertebralardaki hipoplaziler nedeniyle kifoskolyoz ve servikal kifoz oluşur. Tüm tubuler kemikler kalın ve kısadır. Diz eklemleri gevşek, kalça eklemleri sorunludur.
Frontonazal displazi, otosomal resesif yolla aktarılan, alın ve burun bölgelerinin malformasyonlarıyla karakterize kalıtsal bir sendromdur; ALX3 genindeki mutasyonun sonucudur. En önemli bulgu, frontal kemikte ve yüz kemiklerinde orta çizgi yarıklarının bulunmasıdır ile bunun sonucunda ortaya çıkan nazofrontal ensefalosel’dir. Alın derisinde lipoma olabilir. Mikroftalmi saptanır, gözler birbirinden aşırı uzaktadır (hipertelorizm); ptozis, kapak bileşkelerinde anomaliler, katarakt ve epibulbar dermoidler belirlenir. Kulak kepçeleri aşağıdadır, işitme sorunları vardır. Burun kökü aşırı yayvandır; bu alanda saptanan malformasyonların en önemlisi yarıktır. Üstçenede, paranazal sinüslerde ve frontal sinüslerde hipoplazi izlenir. Kalpte Fallot tetralojisi saptanabilir. Pektoral kaslar hipoplazi ya da agenez olabilir. El parmakları kısa ve kıvrıktır. Beyinde Corpus callosum anomalileri saptanır, zeka geriliği vardır. Akromelik frontonazal disostoz fenotipindeki bulgular daha azdır.

Kampomelik displazi, Kamptomelik displazi, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Cüce kısalığına varabilen bacak kısalığı ve genel gelişme geriliği vardır. Kafatası iri (makrosefali), alın geniştir. Hipertelorizm saptanır. İşitme sorunları olabilir. Burun sırtı yayvandır.
Kniest displazi, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Cücelik düzeyine ulaşabilen gelişme geriliği saptanır. Boyun kısadır. Birbirlerinden uzakta fırlak gözlerde (hipertelorizm) myopi, retina dekolmanı ve katarakt saptanır. Sık sık ortaya çıkan otitis media nedenli işitme sorunları olabilir. Yüz yuvarlak ve basıktır. Yarık damak yakınması vardır. Epifiz kıkırdakları geç kapanır, epifizler iridir (megaepifiz) ve ağrılı eklemler hareketleri kısıtlar. İskelet sisteminde, vertebra anomalileri ve kifoskolyoz, pelvis anomalileri ile ayak parmakları anomalileri saptanır. Yassı kemikler incedir. Çok sayıda herni bulunabilir.
Koolen-de Vries sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Güçlü zeka geriliği sendromlarının önemli bir fenotipidir. Cüceliğe yakın düzeyde gelişme geriliği vardır. Yüz uzun, alın ve yanaklar geniştir. Kulaklar kıvrımlıdır. Gözlerde irisler soluktur; şaşılık, ptozis ve kapak yapışıklıkları izlenir. Yüksek ve silindirik burun sırtının altında armut biçiminde bir burun ucu vardır. Alt dudak sarkıktır. Dar ve çukur damakta çoğu hastada yarık da saptanır. Dişler küçüktür (mikrodonti). Kardiyovasüler sistemde, kalpte septum defektleri, pulmoner kapak stenozu ve aorta anomalileri belirlenir. Meme uçları birbirinden uzaktır. Hidronefroz, üriner sistemde sık görülen bir patolojidir.

Meier-Gorlin sendromu, 8 fenotipi olan, fenotip 6 dışındaki tüm fenotipleri otosomal resesif yolla, fenotip 6 otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Öteki fenotiplere kıyasla, fenotip 1 en sık görülen tipidir.

Multipl pterygium sendromu (Escobar sendromu), otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Lethal türünde fetüsün yaşama yetisi yoktur.

Sipondilokarpotarsal sinoztozis sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. İskelet sisteminin tümünü ilgilendiren anomalilerin yanı sıra, özellikle gövde ve omurga sisteminin gelişmesindeki gerilik nedeniyle kısa boy ve orantısız bir vücut yapısı vardır. Parmak eklemlerinde kaynaşmalar görülür. Skapula, sternum ve kaburga anomalileri izlenir; çok sayıdaki vertebta anomalileri nedeniyle skolyoz ve lordoz saptanır. Toraksı olumsuz biçimde etkileyen çevre kemik anomalilerinin yol açtığı restriktif akciğer hastalığına bağlı solunum sorunları yaşarlar. Uzun kemiklerin epifizleri gelişememiştir ve eklemleri gevşektir. Dirsek eklemi hareketleri çok kısıtlıdır.
Seckel sendromu, 9 fenotipi olan, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Doğumdan önce belirlenen ve yaşam boyu süren, cücelik düzeyinde gelişme geriliği ile kuş kafasını andıran yüz görünümü, mikrosefali ve zeka geriliği saptanır. Alın dardır ve geriye çekilmiş gibidir. Gözler fırlaktır; iki taraflı optik atrofi nedeniyle görme sorunları yaşarlar; gözler çekiktir, kapaklarında yapışıklıklar vardır. Stabismus olabilir. Burun kuş gagasını çağrıştırır. Kulaklar biçimsizdir, kulak memesi yoktur ve işitme güçlüğü belirlenir. Asimetrik bir yüzde, altçene küçüktür ve geridedir (mikroretrognati), damak çukurdur ve yarık saptanır. Dişler küçüktür (mikrodonti) ve eksiktir (hipodonti); bu nedenlerle, çiğneme düzlemi bozuktur (maloklüzyon). Mine hipoplazisi belirgindir.

Bohring-Opitz sendromu, otosomal dominant yolla aktarılan, gelişme ve zeka geriliği bulgularının ön planda olduğu, hastaların çoğunun çocukluk yaşlarında kaybedildiği bir sendromdur. C sendromu'nun fenotipi olarak benimsenir.

Craniofrontonasal sendrom, X-kromozomu aracılığıyla dominant (XLD) olarak aktarılan kalıtsal bir sendromdur.

Genitopatellar sendrom, otosomal dominant yolla aktarılan, kalıtsal bir sendromdur. Mikrosefali ve mikrognati vardır. Saçlar seyrektir. Çekik ve iri gözler, iri bir burun içeren kaba yüz yapısı saptanır. İşitme sorunlarının yanı sıra yarık damak ve dişlerin sürmesinde aksamaları olduğu görülür.
Osteopathia striata, kemik yoğunlaşmasının (osteoskleroz) saptandığı, X-kromozomu aracılığıyla (XLD) aktarılan kalıtsal bir sendromdur; olguların 1/3’ü sporadiktir.

Asfiksiyan torasik displazi sendromu, otosomal resesif yolla aktarılan, 22 fenotipi olan kalıtsal bir sendromdur. İlk tanımlanan fenotip 1, Jeune sendromu olarak bilinmemektedir. Fenotiplerden ikisi Ellis-van Creveld sendromu kapsamındadır. Saldino-Noonan sendromu, Majewski sendromu, Mainzer-Saldino sendromu ve Beemer-Langer sendromu görece sık rastlanan fenotiplerdir.

Wolf-Hirschhorn sendromu, genel gelişme geriliği, kafa ve yüz malformasyonları ve epileptiform atakların görüldüğü, “4. kromozomun kısa kolunun distal kısmında delesyon (4p-)” olarak bilinen kromozom anomalisi sonucu ortaya çıkan izole olgulardır. Kız hastalar görece sıktır; 1/3’ü iki yaşına ulaşamadan kaybedilir.
