
Jacobsen sendromu kromozom anomalisi kökenlidir. Kranyumda trigonosefali ve makrosefali saptanır. Hipertelorizm, kaş ve kirpik anomalileri, kikroftalmi, mikrokornea, epikantus, strabismus, ptozis ve koloboma varlığı önemli göz bulgularıdır. Yüz incelemelerinde biçimsiz kulaklar, küçük burun, mikrognati, ince üst dudak ve balık ağzı görünümü dikkat çekicidir. Konjenital kalp defektleri ve bağışıklık sistemi sorunlarına bağlı yineleyen solunum sistemi infeksiyonları sık görülür. Dış genital organlardaki hipoplaziler ile psikomotor gerilik ve otizm başkaca bulgulardır.

DiGeorge sendromu, 22q11 deletion içeren sendromlar grubunun bir fenotipidir. Olguların bir bölümü kalıtsaldır, otosomal dominant yolla aktarılır.Teratojenlerin ve diabetes mellitus'un neden olduğu mutasyonların da etkisi önemsenmektedir. Gebeliğin 4-7. haftasında beliren 3.-4. faringeal ark kompleksi etkilenmesine bağlı konjenital anomalilerle karakterizedir.

Ellis-van Creveld sendromu, ektodermal displazi bulguları içeren, otosomal resesif geçen kalıtsal bir sendromdur. Asfiksiyan torasik displazi sendromu'nun 22 fenotipinden 2'si Ellis-van Creveld sendromu olarak değerlendirilir.
GAPO sendromu, ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur. Hastalarda genel bir gelişme geriliği vardır. Alın bombesi yüksektir. Saçlar giderek dökülür. Optik atrofi ve glokomun neden olduğu görme sorunları ile göz titremeleri (nistagmus) saptanır.
Hallermann-Streiff sendromu , genellikle spontan gen mutasyonu sonucu ortaya çıkan bir sendromdur. Doğum kilosu düşüktür. Yaşam boyu süren genel gelişme eksikliği cücelikle sonlanır.
LADD sendromu; lacrimo-auriculo-dento-digital sendrom; Levy-Hollister sendromu), ektodermal displazi bulguları da içeren, gözyaşı bezi,kulak, diş ve parmak bulgularının baskın olduğu, otosomal dominant yolla geçen kalıtsal bir sendromdur.
Mikrognati (micrognathism), altçenenin yetersiz gelişmesi (hipoplazi) niteliğinde bir anomalidir; 1. ve 2. faringeal arklara (brankial yarık) özgü malformasyonların çoğu çene-yüz bölgesindeki mezenkimal dokunun embriyolojik dönemdeki yetersizliğine bağlanmaktadır. Altçene küçüktür ve geridedir; bu olgu, çevredeki kasların hareketlerini sınırlarken bu kaslardan yararlanan dokuların gelişmeleri de kısıtlanır. Kraniyofasiyal anomalileri içeren sendromlarda çok sık görülen bulgulardan biridir. Sendroma-özgü olmayan olguların varlığı da bilinmektedir; örneğin; altçenesi, kafa ile göğüs kafesi arasında sıkışmış olan bir fetüste çene kemiklerinin gelişimi olması gereken düzeye ulaşamamaktadır. Gebelik sorunları, gebelerin alkol kullanması, gestasyonel diabet (gebelik diabeti) gibi çevresel faktörler etkili olabilmektedir (Möbius sendromu). Olguların bir bömümü, gebelerdeki rubella (kızamıkçık) infeksiyonu komplikasyonudur. Yenidoğanların bir bölümünde görülebilen hafif mikrognatiler, altçene gelişiminin tamalanmasıyla silinebilir. Mikrognati'de altçene gövdesi kadar çene eklemini oluşturan yapıların hipoplazisi de önemlidir. Mikrognatilerin bir bölümü kulak sistemi, üstçene ve damak gelişiminin de aksadığı olgularla birlikte görülür.
TORCH, annedeki bir infeksiyon etkeninin, plasenta aracılığıyla ya da doğum sırasında bebeğe geçmesi olgusudur. TORCH tablosu ölü doğum, erken doğum ve prematüre bebek ölümlerinin önemli nedenlerinden biridir. İnfeksiyon etkenlerinin baş harfleriyle oluşturulan TORCH kısaltmasında yer alan canlı etkenlere giderek yenileri eklenmektedir.
Konjenital hemihipertrofi ya da konjenital hemihiperplazi, otosomal dominant yolla aktarılan, Beckwith-Wiedemann sendromunun fenotipi olabilen, aşırı büyüme sendromlarına özgü bulgular içeren kalıtsal bir sendromdur. Gen mutasyonuna bağlı izole olgular görülebilir.
Möbius sendromu, maksillofasiyal, lokomotor ve nörolojik bulguların ön planda olduğu bir sendromdur. Olguların önemli bir bölümünü izole olgulardır; etyolojilerinde çevresel faktörlerin etkisi belirlenir. Otosomal dominant yolla aktarılan kalıtsal olgulara ancak birkaç ailede rastlanılmıştır. Oromandibular-limb hipogenezi grubu içinde yer alır. Yüzün bir yarısını etkileyen (unilateral) olgulara da rastlanmıştır.
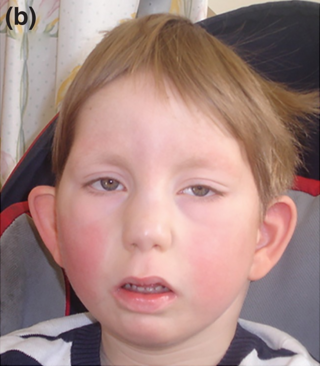
Au-Kline sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; böbrek, kalp ve yüz bulgularıyla öne çıkar.
Yarık damak-lateral sineşi sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; izole olgular çok enderdir. Yenidoğan bulgularının başında maksilla ile mandibula kemiklerinin konjenital yapışıklığı (syngnathia) ile dilin bantlarla, yan kenarlarından, damağa ve ağız tabanına yapışıklığı gelir. Üstçene küçüktür (mikrognati) ve yarık saptanır. Alt dudak çıkıntılılıdır, dudak kommisuraları aşağı dönüktür. Filtrum kısa, yanaklar geniştir. Gözyaşı kanalı tıkanıklığı, inguinal fıtıklar, zeka geriliği olası öteki bulgulardır.

Joubert sendromu, kalıtsal bir sendromdur; 36 fenotipinden 34'ü otosomal resesif yolla aktarılır, olguların bir bölümü Meckel sendromu ile çakışmaktadır. Joubert sendromunun tüm fenotiplerinde 3 temel bulgu vardır: santral sinir sistemi anomalileri, fizik gelişme geriliği, solunum sorunları. Serebellum (beyincik) vermisi ve beyin sapı hipoplazisi ile beyin radyolojisinde saptanan “azıdişi bulgusu ” santral sinir sistemi anomalilerinin başında gelir. Ataksi ve hipotoni sık görülür. Bazı hastalarda oksipital meningosel ya da meningomyelosel olabilir. Schinzel acrocallosal sendromu, önemli fenotiplerinden biridir.

Kabuki sendromu, iki fenotipi olan bir sendromdur: Kabuki sendromu 1 ve Kabuki sendromu 2 (Kabuk2). Kabuk1, otosomal dominant yolla aktarılan kalıtsal ya da izole olgular biçiminde görülür; Kabuk2 ise, X-kromozomu aracılığıyla dominant yolla (XLD) aktarılır. Her iki fenotipini çok sayıda ortak bulguları vardır; özellikle yüz, iskelet sistemi, gelişme geriliği, parmakizleriyle ilgili bulgular ve zeka geriliği önemlidir.

Jaffe-Campanacci sendromu, gen mutasyonuyla ortaya çıkan izole sendromlardandır. Deride café-au-lait pigmentasyonu, uzun kemiklerde non-ossifying fibromalar ile bunların neden olduğu patolojik kırıklar, çenelerde dev hücreli reparatif granülom özelliği taşıyan lezyonlar, hipogonadizm ve zeka geriliği bulgularını içerir. Kalıtsal olgular az sayıdadır ve Nörofibromatoz tip 1 grubu içinde yer alırlar.

Goltz sendromu, X kromozomu aracılığıyla dominant (XLD) olarak aktarılan kalıtsal bir sendromdur; erkek fetüslerin çok büyük bölümü intrauterin evrede (rahimde) öldüğü için hastaların çoğu kız bebeklerdir.
Aşırı büyüme sendromları , çocuklarda, doğum öncesi (prenatal) dönemde ya da doğumdan sonra (postnatal) dönemde ortaya çıkabilen, bebeklerin/çocukların vücutlarının tümünde ya da bir bölümünde ortaya çıkan irileşmeyle karakterize olgulardır; aşırı büyümelerin görüldüğü sendromlarda, kilo ve boy artışı, çeşitli anomaliler ile zeka geriliği ve tümör riskinin varlığı en sık rastlanan bulgulardır.

Multipl pterygium sendromu (Escobar sendromu), otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Lethal türünde fetüsün yaşama yetisi yoktur.

Hartsfield sendromu, otosomal dominant yolla aktarılan, ender görülen, kalıtsal bir sendromdur. El ve ayak parmaklarının konjenital eksikliği (ektrodaktili), yarık dudak ve yarık damak ile kafatası malformasyonu (holoprosensefali) üçlüsü temel bulgulardır.

Klippel-Feil anomalisi, boyun omurlarını ilgilendiren konjenital bir anomalidir; kalıtsal olgular otosomal dominant yolla aktarılır. Klippel-Feil sendromu'nun temel anomalileridir. Başlıca 3 bulgu saptanır:
- Servikal vertebralarda hipoplazi ve kaynaşma
- Servikal vertebralardaki anomaliye bağlı olarak boyun kısa ve hareketleri sınırlıdır
- Trapez kaslarında(*) yelkensi görünüm saptanır.