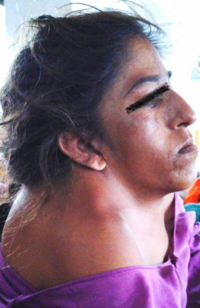
Goldenhar sendromu, Craniofacial microsomia sendromu'nun otosomal dominant yolla aktarılan kalıtsal nitelikteki fenotipidir. Otosomal dominant yolla aktarılan en önemli kalıtsal örnektir.

Chiari malformasyonları, serebellum’daki ve medulla spinalis’teki yapısal defektlerin ön planda olduğu bir grup patolojiden oluşur; beyin sapının alt bölümündeki oluşum kusurlarının sonucu ortaya çıkar. Bu bölgedeki yetersiz gelişme kafatası boyutlarının da küçük kalmasına neden olur.
Ağız-Yüz-Parmak sendromu , günümüze dek 16 fenotipi belirlenmiş olan bir sendromlar kümesidir. Bilinen fenotiplere yenileri eklenebilir. Ağız-Yüz-Parmak sendromunun OFD arasında en sık ratlanılanı OFP tip I temel bulguları içerir; sıkça rastlanan öteki fenotiplerde, tip I'e eklenen ya da çıkarılan yan bulgular vardır.
OPD sendromu (otopalatodigital sendrom), X-kromozomu aracılığıyla dominant (XLD) yolla geçen kalıtsal bir sendromdur (Gorlin-Cohen sendromu hariç). Kız çocukları daha hafif etkilenir. Tümü benzer bulgular içeren 5 tipi vardır:
- Otopalatodigital sendrom tip I (OPD tip1)
- Otopalatodigital sendrom tip II (OPD tip2)
- Gorlin-Cohen sendromu (ilgili sayfaya gidiniz)
- Melnick-Needles sendromu (tip 1 ile allelik bağlantısı vardır; ilgili sayfaya gidiniz)
- Terminal osseöz displazi ve pigmentli deri defektleri (çok enderdir, ayrıntı verilmemiştir).
Gorlin-Cohen sendromu , ektodermal displazi bulgularını da içerebilen bir OPD sendromu tipidir. 2 fenotipi vardır:
- Frontometaphyseal dysplasia 1
- Frontometaphyseal dysplasia 2
Auriculocondylar sendrom, kalıtsal bir sendromdur. 3 fenotipi vardır. Fenotip 1 otosomal dominant (AD), fenotip 3 otosomal resesif (AR), fenotip 2 ise her iki yolla aktarılır. 1. ve 2. Fenotiplerin bulguları ortaktır; etkilenen gen farklıdır.
Aase-Smith sendromu, otosomal dominant yolla aktarılan, 2 fenotipi olan, kalıtsal bir sendromdur. Fenotip 2, Diamond-Blackfan anemisi grubunun 6. üyesidir.

Cornelia de Lange sendromu , 5 fenotipi olan kalıtsal bir sendromdur. Tüm fenotiplerde benzer bulgular saptanır; farklılık etkilenen genlerdedir.
Frontonazal displazi, otosomal resesif yolla aktarılan, alın ve burun bölgelerinin malformasyonlarıyla karakterize kalıtsal bir sendromdur; ALX3 genindeki mutasyonun sonucudur. En önemli bulgu, frontal kemikte ve yüz kemiklerinde orta çizgi yarıklarının bulunmasıdır ile bunun sonucunda ortaya çıkan nazofrontal ensefalosel’dir. Alın derisinde lipoma olabilir. Mikroftalmi saptanır, gözler birbirinden aşırı uzaktadır (hipertelorizm); ptozis, kapak bileşkelerinde anomaliler, katarakt ve epibulbar dermoidler belirlenir. Kulak kepçeleri aşağıdadır, işitme sorunları vardır. Burun kökü aşırı yayvandır; bu alanda saptanan malformasyonların en önemlisi yarıktır. Üstçenede, paranazal sinüslerde ve frontal sinüslerde hipoplazi izlenir. Kalpte Fallot tetralojisi saptanabilir. Pektoral kaslar hipoplazi ya da agenez olabilir. El parmakları kısa ve kıvrıktır. Beyinde Corpus callosum anomalileri saptanır, zeka geriliği vardır. Akromelik frontonazal disostoz fenotipindeki bulgular daha azdır.

Robinow sendromu, 5 fenotipi olan kalıtsal bir sendromdur; fenotiplerin ikisi otosomal resessif, üçü otosomal dominant yolla aktarılır. Tüm fenotiplerde iskelet sistemi bulguları yoğun olmakla birlikte maksillofasiyal bulguları ile her iki cinste de saptanan genital organ hipoplazileri ortak bulgulardır.

Meier-Gorlin sendromu, 8 fenotipi olan, fenotip 6 dışındaki tüm fenotipleri otosomal resesif yolla, fenotip 6 otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Öteki fenotiplere kıyasla, fenotip 1 en sık görülen tipidir.

Bohring-Opitz sendromu, otosomal dominant yolla aktarılan, gelişme ve zeka geriliği bulgularının ön planda olduğu, hastaların çoğunun çocukluk yaşlarında kaybedildiği bir sendromdur. C sendromu'nun fenotipi olarak benimsenir.

Fraser sendromu, otosomal resesif yolla aktarılan, 3 fenotipi olan, kalıtsal bir sendromdur; göz kapaklarının birbirlerine yapışık olması (kriptoftalmi), yapışık parmaklar (sindaktili) ile solunum-üriner-genital sistemlerin anomalileri başlıca bulgulardır.

Meckel-Gruber sendromu, çok sayıda genin etkilenmesinden kaynaklanan, bu nedenle 15 fenotipi olan, otosomal resesif yolla aktarılan, siliyopatiler grubundan, kalıtsal bir sendromdur. Kistik böbrekler, beyin anomalileri ve ensefalosel, polidaktili ve karaciğer anomalileri başlıca bulgulardır. En sık görülen fenotip Meckel sendromu 1, öteki fenotiplerdeki bulguların çok büyük bir bölümünü içerir. Bazı fenotipleri Joubert sendromuyla çakışan nitelikleri içermektedir.

Klippel-Feil anomalisi, boyun omurlarını ilgilendiren konjenital bir anomalidir; kalıtsal olgular otosomal dominant yolla aktarılır. Klippel-Feil sendromu'nun temel anomalileridir. Başlıca 3 bulgu saptanır:
- Servikal vertebralarda hipoplazi ve kaynaşma
- Servikal vertebralardaki anomaliye bağlı olarak boyun kısa ve hareketleri sınırlıdır
- Trapez kaslarında(*) yelkensi görünüm saptanır.

Saethre-Chotzen sendromu (acrocephalosyndactylia III), fiziksel gelişme geriliği, kraniyosinostoz nedenli kafatası anomalileri, asimetrik yüz, göz ve parmak malformasyonlarının saptandığı otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Kraniyosinostozun çok sayıda eklemi etkilediği olgularda “kafaiçi basıncı artışı sendromu (KİBAS)” gelişebilir.

Shprintzen-Goldberg sendromu , marfanoid yapı, kraniyosinostoz, kardiyovasküler ve nörolojik anomalilerle karakterize, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Marfanoid yapı, Marfan sendromundaki bulguların büyük bölümünün saptandığı, ancak Marfan sendromunudaki genetik altyapının bulunmadığı olgular için kullanılan nitelemedir; uzun kollar ve bacaklar, örümceksi parmaklar (araknodaktili), eklemlerde aşırı gevşeklik ve mitral kapak prolapsusu Marfan sendromunu andıran başlıca bulgulardır. Robbins Shprintzen-Goldberg sendromu’nun, Marfan sendromu ve Loeys-Dietz sendromu ile oldukça çok büyük benzerlikleri vardır.
BOR sendromu, otosomal dominant aktarılan kalıtsal bir sendromdur.
Otofasiyoservikal sendrom (otofaciocervical sendrom), yüz ve iskelet malformasyonlarının ön planda olduğu, 2 fenotipi saptanan bir sendromdur. Fenotiplerde genler farklıdır; biri otosomal dominant, öteki otosomal resesif yolla aktarılır. BOR sendromu ile aleldir.
Meckel sendromu, birden fazla etkisi olan bir genle ilgili (pleiotropic), otosomal resesif yolla aktarılan, 15 kadar fenotipi bulunan kalıtsal bir sendromdur. Çok sayıda ve karmaşık bulguların varlığı tanıda güçlükler oluşturur. Bazı uzmanlar, Meckel sendromunun 3 temel bulgusu olduğunu savunurlar: (i) Santral sinir sistemi malformasyonları ; (ii) Kistik böbrekler; (iii) Karaciğer patolojileri. Bebeklerin yaşama yetisi çok kısıtlıdır; canlı doğabilenlerin çoğu birkaç hafta içimde yitirilir.