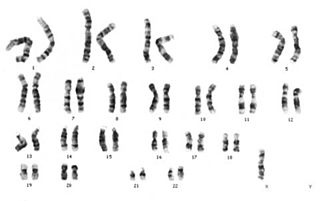
Klinefelter sendromu

Klinefelter sendromu ya da 47, XXY sendromu; hücre bölünmesi sırasında, eşeysel kromozom düzensizliklerinden kaynaklanan semptomların kişide görülmesi durumudur.
1942'de Dr. Harry Klinefelter ilk olarak tanımlamıştır, doktorun adıyla anılan doğuştan gelen bir özelliktir.
Nedenleri
Hücre bölünmesi sırasında eşey kromozomlarından X'in ayrılmaması durumundan kaynaklanan bir sendromdur. İki tane X kromozomu taşıyan bir yumurta hücresinin normal bir sperm ile döllenmesiyle meydana gelir. Normal karyotipte 46, XY olması gereken bireyin, Klinefelter sendromunda 47, XXY şeklinde karyotipi vardır.
Görünümleri
Bu durumdaki kişiler genellikle erkek birey olarak görülür. Puberteyle ortaya çıkar (puberte gecikir). Uzun kol ve bacakları, kadınımsı kalça çıkıntıları ilk olarak göze çarpan özellikleridir. Testisleri küçük, kadınımsı göğüs (jinekomasti) ve kas gelişimleri vardır. Sesleri erkeklere nazaran daha incedir. Hipogonadizm görülebilir. Sakal ve bıyık gelişimleri çok az, vücut kıllanmaları kadınımsı görünümdedir. Spermatogenez görülmez ve kısır bireylerdir.[1]
Canlı erkek doğan bireylerin 500 ya da 1000'inde 1 oranla görülür.
- Mikrosefali
- Silindirik yüz
- Prognatizm (alt çene/üst çene)
- Yarık damak
- Maloklüzyon
- Periodontal patolojiler
- Taurodontism
- Prostat küçük
- Penis normal/hafif küçük
- Varisler
- Ekstragonadal germ hücreli tümör riski (pineal doku, presakral bölge, mesane, prostat, karaciğer)
- Meme kanseri riski
- Psikomotor gelişme geriliği
- Testosteron düzeyi düşük
Türleri

48, XXYY sendromu; yaklaşık 17,000 canlı doğumda bir karşılaşılan Klinefelter sendromunun özelliklerini taşıyan bir hastalıktır. Bu hastalarda semptomlar daha ağır görülür.
Mozaik 47,XXY/46,XY Klinefelter sendromu; şeklinde mozaik olarak da görülebilir. Bu hastalarda semptomlar 47,XXY karyotipli hücrelerin yoğunluğuna bağlı olarak değişir.
Doğum öncesi tanı
Klinefelter sendromu gebelikte tanınabilen bir hastalıktır. Nadir olarak, ultrason bulguları ile fetüs anormal olarak tanımlanabilir. Amniyosentez ve diğer bazı tanı yöntemleri ile Klinefelter sendromununa gebeliklerde kesin tanı konur.
Tedavi
Son yıllarda testis dokusundan sperm elde etme ve mikroenjeksiyon (ICSI) yöntemleri ile Klinefelter Sendromlu erkeklerin çocuk sahibi olmaları sağlandı.[5] Klinefelter sendromu erkeklerde yapılan geniş serilerde testisten sperm elde etme oranı mikro-TESE yöntemi ile yaklaşık %50 oranında olduğu gösterilmiştir.[6][7] Testisten sperm elde olasılığının 35 yaşının altında olan Klinefelter sendromlu erkeklerde daha ileri yaştaki erkeklere oranla daha yüksek olduğu saptandı.[6] Testisten elde edilen spermlerin ile eşlerinin yumurtalarına mikroenjeksiyon yöntemi ile aktarılması yoluyla bu erkeklerin çocuk sahibi olma olasılığının testisten sperm elde edilen ve genetik bozukluğu olmayan erkeklerden farklı olmadığı gösterilmiştir.[7] Klinefelter sendromlu erkeklerden alınan spermlerle yapılan mikroenjeksiyon tedavileri sonrasında doğan çocuklarda genetik anomali görülmemiştir.[7]
Kılavuz
Eylül 2020, Avrupa Androloji Akademisi ilk kez Klinefelter sendromu için kılavuzlar yayınladı.[8]
Ayrıca bakınız
- Turner sendromu
- XYY sendromu
- Triple X sendromu
- 48,XXYY sendromu
Dış bağlantılar
- Ulusal Çocuk Sağlığı Enstitüsü2 Eylül 2006 tarihinde Wayback Machine sitesinde arşivlendi.
- XXYTalk Klinefelter Sendromlular Topluluğu6 Eylül 2008 tarihinde Wayback Machine sitesinde arşivlendi.
- Klinefelter Sendromu ve Dernekleri28 Ekim 2006 tarihinde Wayback Machine sitesinde arşivlendi.
- Amerikan Klinefelter Sendromlular Derneği, Bilgi ve Destek2 Mayıs 2020 tarihinde Wayback Machine sitesinde arşivlendi.
- 47xxy.org4 Ekim 2006 tarihinde Wayback Machine sitesinde arşivlendi.
- Kkinefeltersyndrome.org9 Kasım 2006 tarihinde Wayback Machine sitesinde arşivlendi.
- XXYY Sendromu ve XXYY Projesi7 Ocak 2014 tarihinde Wayback Machine sitesinde arşivlendi.
Kaynakça
- ^ Visootsak J, Aylstock M, Graham JM Jr. Klinefelter syndrome and its variants: an update and review for the primary pediatrician. Clinical Pediatrics (Philadelphia), 40(12):639-651, 2001
- ^ Groth KA, Skakkebæk A, Høst C, et al. Clinical review: Klinefelter syndrome - a clinical update. The Journal of Clinical Endocrinology and Metabolism, 98(1):20-30, 2013
- ^ Haritha A, Jayakumar A. Syndromes as they relate to periodontal disease. Periodontology 2000, 56:65–86, 2011
- ^ Bonomi M, Rochira V, Pasquali D, et al. Klinefelter syndrome (KS): genetics, clinical phenotype and hypogonadism. Journal of Endocrinological Investigation, 40(2):123-134, 2017
- ^ Ron-el, R.; Friedler, S.; Strassburger, D.; Komarovsky, D.; Schachter, M.; Raziel, A. (Şubat 1999). "Birth of a healthy neonate following the intracytoplasmic injection of testicular spermatozoa from a patient with Klinefelter's syndrome". Human Reproduction (Oxford, England). 14 (2): 368-370. doi:10.1093/humrep/14.2.368. ISSN 0268-1161. PMID 10099981. 16 Aralık 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 16 Aralık 2021.
- ^ a b Emre Bakircioglu, Mustafa; Erden, Halit Firat; Kaplancan, Tansel; Ciray, Nadir; Bener, Faruk; Bahceci, Mustafa (Kasım 2006). "Aging may adversely affect testicular sperm recovery in patients with Klinefelter syndrome". Urology. 68 (5): 1082-1086. doi:10.1016/j.urology.2006.05.028. ISSN 1527-9995. PMID 17095066. 16 Aralık 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 16 Aralık 2021.
- ^ a b c Bakircioglu, Mustafa Emre; Ulug, Ulun; Erden, Halit Firat; Tosun, Suleyman; Bayram, Asina; Ciray, Nadir; Bahceci, Mustafa (Nisan 2011). "Klinefelter syndrome: does it confer a bad prognosis in treatment of nonobstructive azoospermia?". Fertility and Sterility. 95 (5): 1696-1699. doi:10.1016/j.fertnstert.2011.01.005. ISSN 1556-5653. PMID 21295296. 16 Aralık 2021 tarihinde kaynağından arşivlendi. Erişim tarihi: 16 Aralık 2021.
- ^ Zitzmann, Michael; Aksglaede, Lise; Corona, Giovanni; Isidori, Andrea M.; Juul, Anders; T'Sjoen, Guy; Kliesch, Sabine; D'Hauwers, Kathleen; Toppari, Jorma; Słowikowska‐Hilczer, Jolanta; Tüttelmann, Frank (6 Ekim 2020). "European academy of andrology guidelines on Klinefelter Syndrome: Endorsing Organization: European Society of Endocrinology". Andrology (İngilizce): andr.12909. doi:10.1111/andr.12909. ISSN 2047-2919. 27 Ekim 2020 tarihinde kaynağından arşivlendi. Erişim tarihi: 24 Ekim 2020.