
Otizm, üç yaşından önce başlayan ve ömür boyu süren, sosyal etkileşime ve iletişime zarar veren, sınırlı ve tekrarlanan davranışlara yol açan beynin gelişimini engelleyen bir rahatsızlıktır. Bu belirtiler otizmi, Asperger sendromu gibi daha hafif seyreden otistik spektrum bozukluğundan (OSB) ayırır. Otizm kalıtımsal kökenlidir ancak kalıtsallığı oldukça karmaşıktır ve OSB'nin kökeninin çoklu gen etkileşimlerinden mi yoksa ender görülen mutasyonlardan mı kaynaklandığı çok açık değildir. Nadir vakalarda, doğum sakatlıklarına neden olan etmenlerle yakından bağlantılıdır. Diğer görüşlere göre ise çocuklukta yapılan aşılar gibi nedenler tartışmalıdır ve aşı kökenli varsayımların ikna edici bilimsel kanıtları yoktur. 2007 yılında yapılan araştırmalara göre otizmin prevalansını 1.000 kişiye bir ya da iki vaka olarak tahmin eder, aynı araştırmalardaki tahminlere göre OSB yaklaşık 1.000 kişide altı vakadır ve erkeklerde rastlanma oranı kadınlara göre 4,3 kat daha fazladır. 2022 yılı CDC verilerine göre otizmin görülme sıklığı 44 çocuktan 1'e yükselmiştir. Otizm vakalarının sayısı 1980'lerden beri oldukça fazla oranda artmıştır. Bunun nedeni kısmen tanı koyma yöntemlerindeki değişikliklerdir; gerçek prevalansın artıp artmadığı anlaşılamamıştır.

Down sendromu, trizomi 21 ya da mongolizm; genetik düzensizlik sonucu insanın 21. kromozom çiftinde fazladan bir kromozom bulunması durumu ve bunun sonucu olarak ortaya çıkan genetik bir bozukluktur. Down sendromu, bireyin 1 yaşından daha uzun süre yaşayabildiği tek otozomal trizomidir.

Noonan sendromu kalıtsal bir sendromdur; bazı uzmanlarca Turner sendromu'nun erkek çocuklarında görülen tipi olarak tanımlanır. 10 fenotipi vardır, bunlardan fenotip 2 otosomal resessif, öteki 9 fenotip otosomal dominant kalıtım niteliği taşır. Hem çocukları hem de yetişkinleri etkiliyen bir hastalıktır. Genellikle genetik kalp rahatsızlıkları ve iskelet bozuklukları ile ilişkilendirilmektedir.

Kedi gözü sendromu , ebeveynlerden birinde spontan olarak ortaya çıkan kromozom anomalisinin otosomal dominant yolla aktarıldığı izole bir sendromdur. Kromozomları normal olan bireylerin 2 adet 22p olarak bilinen kısa kolu ve 22q olarak adlandırılan 2 adet uzut kolu olan 2 tane kromozom 22'leri vardır. Fakat, kedi gözü sendromuna sahip bireylerde ise kromozom 22'nin kısa kolu ve uzun kolunun bir bölümü vücudun hücrelerinde ikişer adet bulunacaklarına üç ya da dört adet bulunurlar. Fetüslerin çoğu ölü doğar. "Kedi gözü sendromu" adı hasta bireydeki belirgin göz anomalisinden türetilmiş bir isimdir.
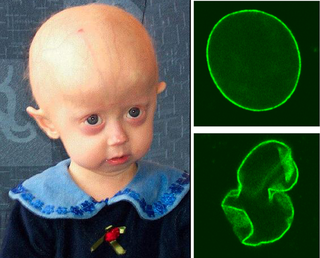
Progeria, halk dilindeki adıyla erken yaşlanma hastalığı, konu hakkında yapılan bilimsel araştırmalar hastalığın çaresini bulmaktan ziyade hastalığa sebep olan faktörleri bulmak ve bu sayede insanlığın ömrünü uzatabilmektir.
Asperger sendromu (AS) ya da Asperger bozukluğu, sosyal etkileşimde zorluklar ve sınırlı, basmakalıp ilgi ve etkinliklerle tanımlanan otistik spektrum bozukluklarından (OSB) biridir. AS diğer OSB’lerden dil ve bilişsel gelişimde genel bir gecikme olmamasıyla ayrılır. Her ne kadar standart tanı ölçütleri arasında belirtilmemişse de motor sakarlık ve sıra dışı dil kullanımına sıklıkla rastlanır.
Rett sendromu, yaygın gelişimsel bozukluklardan birisi olarak sınıflandırılan beyinsel gelişim bozukluğudur. Ancak bunun yanlış bir sınıflandırma olduğunu ve benzer şekilde otistik belirtiler gösteren frajil X sendromu, tüberoz skleroz ya da Down sendromunun yaygın gelişimsel bozukluklar olarak sınıflandırılabileceğini önesüren görüşler bulunmaktadır. Bu sendromun belirtileri kolaylıkla otizm ve Angelman sendromunun belirtileriyle karışır. Klinik belirtiler arasında baş büyüme hızının azalması ve bazen mikrosefali, küçük el ve ayaklar bulunur. Stereotipik ve yineleyici el hareketleri de gözlenir. Bilişsel bozukluk ve gerileme döneminde de sosyalleşme sorunları da belirtiler arasında görülür. Okula girdikleri dönemde sosyalleşme genellikle düzelir. Rett sendromu olan kız çocuklar gastrointestinal bozukluklara yakalanmaya yatkındır ve %80’i nöbet geçirir. Hemen hemen hiç sözel becerileri yoktur ve kadınların %50’si yürüyemez. Skolyoz, büyüme eksikliği ve kabızlık çok yaygındır ve sorunlu olabilir.
Angelman sendromu ilk olarak 1965 yılında İngiliz doktor Harry Angelman tarafından tanımlanmış nörogenetik bir bozukluktur. Irklarda görülme hızı çok iyi bilinmemekle beraber yaklaşık ensidansın 15,000 ila 30,000 canlı doğumda bir olduğu kabul edilmektedir. Anneden gelen kromozom 15'teki bir bozukluktan kaynaklandığı sanılmaktadır. Hastalığın temel bulguları zeka geriliği, yürüyüş-koordinasyon bozukluğu, konuşma bozukluğu, konvülsiyon ve uygunsuz gülümsemelerdir. Hatta bu sebeple hastalık bazen “mutlu kukla ” sendromu olarak da bilinir.
Hunter sendromu ya da Tip II mukopolisakkaridoz, iduronat-2-sülfataz (I2S) enziminin eksikliğinden ya da yokluğundan kaynaklanan lizozomal depo hastalığıdır. Doktor Charles A. Hunter (1873-1955) tarafından ilk kez 1917 yılında tanımlandığından, Hunter sendromu olarak adlandırılır.

Aase sendromu veya Aase–Smith sendromu, anemi ile birlikte bazı eklem ve iskelet deformiteleriyle karakterize edilen nadir bir kalıtsal hastalıktır. Aase sendromunun otozomal baskın kalıtsal bir bozukluk olduğu öne sürülmektedir. Hastalığın genetik temeli henüz aydınlatılamamıştır. Anemi, kan hücrelerinin üretildiği kemik iliğinin hipoplazisinden kaynaklanmaktadır.

XXYY sendromu, erkeklerin fazladan bir X ve Y kromozomlarının bulunduğu eşey kromozomları bozukluğu. İnsan hücreleri genellikle anne ve babadan olmak üzere iki cinsiyet kromozomu içerir. Genellikle dişiler iki X kromozomuna (XX), erkekler bir X bir Y kromozomuna (XY) sahiptir. Düzgün çalışan bir SRY geni en az bir Y kromozomunun ortaya çıkmasıyla erkek olur. Bu nedenle XXYY kromozomuna sahip olan insanlar normalde erkek olur. XXYY sendromlu erkekler 46 yerine 48 kromozoma sahiptir. Bu yüzden XXYY sendromu bazen 48, XXYY sendromu olarak yazılır. Yaklaşık her 18.000-40.000 erkek doğumunda bir XXYY sendromlu bireyin doğduğu tahmin edilmektedir.
Ektodermal displazi sendromları, ektodermden gelişen doku ve organlardaki malformasyonların ve oluşum kusurlarının saptandığı geniş bir topluluktur. Dişlerle ilgili anomaliler ve malformasyonlar; saç oluşumunda yetersizlikler (seyrek/kırılgan/oluşmama); ter bezlerinin eksikliğine bağlı terleme azlığı/yokluğu (hipohidroz); başkaca çeşitli deri ve tırnak patolojileri ortak bulgular olarak saptanır. Günümüze dek ektodermal displazi bulgularını içeren 200'e yakın sendrom bildirilmiştir; bu kadar çok sayıda fenotip bulunmasının nedeni farklı genlerde oluşan mutasyonlardır. Ektodermal displazili ailelerdeki kalıtım otosomal dominant, otosomal resesif ya da x-kromozomu aracılığıyla resesif olarak aktarılır.

Sotos sendromu, 3 fenotipi olan aşırı büyüme sendromlarından biridir. Sotos 1, serebral gigantizm olarak da bilinen, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Sotos 2, Malan sendromu olarak da bilinir; olguların çoğu nedensiz (spontan) olarak ortaya çıkar, kalıtsal olan birkaç olgu vardır. Sotos 3, otosomal resesif yolla aktarılan, çok ender görülen kalıtsal bir fenotiptir.

Aarskog-Scott sendromu, X'e bağlı kalıtılan (XLR), nadir görülen bir hastalıktır ve kısa boy, yüz anomalileri, iskelet ve genital anomaliler ile karakterize edilir. Bu sendrom çoğunlukla erkeklerde görülmekle birlikte, kadınlarda da sendromun hafif belirtileri bulunabilir.

Vici sendromu, psikomotor gelişme geriliğinin yanı sıra göz, dolaşım sistemi ve bağışıklık sistemindeki defektlerle karakterize, otosomal resesif yolla aktarılan kalıtsal bir sendromdur.

Limb-mammary sendromu, ektodermal displazi bulguları, el-ayak anomalileri ve memelerdeki oluşum kusurlarıyla karakterize otosomal dominant yolla aktarılan kalıtsal bir sendromdur. TP63 geninin mutasyonu sonucu ortaya çıkan sendromlardandır.

Waardenburg sendromu, en azından bir dereceye kadar doğuştan işitme kaybı ve pigmentasyon eksiklikleri ile karakterize edilen, parlak mavi gözleri, poliosis veya açık ten lekelerini içerebilen bir grup nadir genetik durumdur. Bu temel özellikler, durumun tip 2'sini oluşturur; tip 1, olarak adlandırılan gözlerin iç köşeler arasında daha geniş bir aralık olan telekantus veya distopia kantorum olarak adlandırılan tipi de mevcuttur. Nadir görülen tip 3'te, kollar ve eller de bozuk, kalıcı parmak kontraktürleri veya kaynaşmış parmaklar, tip 4'te ise kişide bağırsak disfonksiyonuna yol açan doğuştan sinir eksikliği olan Hirschsprung hastalığı vardır. Ayrıca, gelişimsel gecikme ve kas tonusu anormallikleri gibi merkezi sinir sistemi semptomlarına neden olabilecek en az iki tip vardır.
Bir örtüşme sendromu, daha yaygın olarak tanınan en az iki bozukluğun özelliklerini paylaşan tıbbi bir durumdur. Örtüşme sendromlarının örnekleri, romatolojide örtüşen bağ dokusu bozuklukları ve kardiyolojide örtüşen genetik bozukluklar gibi birçok tıbbi uzmanlıkta bulunabilir.

Brunner sendromu veya MAO-A eksikliği, MAO-A geninde meydana gelen bir mutasyon sonucu ortaya çıkan çok nadir bir genetik bozukluktur. Ana semptomları ortalamadan düşük IQ seviyesi, impulsif davranışlar, uyku bozuklukları ve sürekli ruh hali değişimleridir.
Albinizm-siyah saç teli-hücre göç bozukluğu, bir bireyin fiziksel görünümünü ve fizyolojisini etkileyen durumları tanımlayan terim ve kavramların kısaltmasıdır: (1) A – albinizm, (2) B – siyah saç teli, (3) C – bağırsak nörositlerinin hücre göç bozukluğu ve (4) D – sinirsel tip işitme kaybı. Bu sendrom, endotelin B reseptör geni (EDNRB) mutasyonundan kaynaklanır.