Kedi miyavlaması sendromu
| Kedi miyavlaması sendromu | |
|---|---|
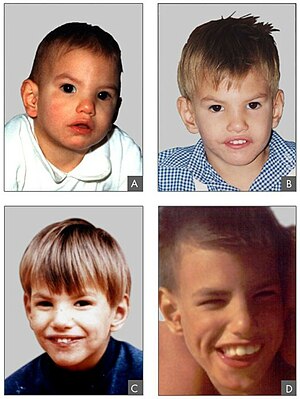 | |
| Kedi miyavlaması sendromlu 8 aylık (A), 2 yaşında (B), 4 yaşında (C) ve 9 yaşındaki (D) hastaların yüz özellikleri | |
| Uzmanlık | Medikal genetik |
Kedi miyavlaması sendromu, Kedi çığlığı sendromu veya tıptaki isimleriyle Cri du Chat sendromu ya da Cri-du-Chat sendromu (5p delesyon sendromu, 5p minus veya Lejeune sendromu olarak da isimlendirilir), 5. kromozomun bir parçasının kaybıyla ilişkili nadir bulunan bir genetik düzensizliktir. Sendromun genetik tanımı 45,X(X/Y),-5p olarak gösterilir. Yani kişide 45 kromozomun bulunduğunu fakat 5. kromozomun kısa (petit) kolunun bir kısmının bulunmadığını ifade eder. Bu tip kromozom mutasyonlarında DNA'daki bazın ya da bazların yok olmasına delesyon adı verilir. Delesyondaki büyüklük bebeklerdeki fiziksel, psikomotor ve zihinsel gelişimlerini etkiler.
İlk olarak Jérôme Lejeune tarafından 1963'te tanımlanmıştır.[1] Durumun etkileri her 20.000 ila 50.000 canlı durumda 1 görülür. Rahatsızlık etnik geçmişi olan bütün insanlarda ve 3/1 oranla dişilerde daha çok görülür.
Belirtiler
Sendrom adını yeni doğanın karakteristik ağlamasından alır. Bebeğin ağlayışı yutak ve sinir sistemindeki sorunlarla ilişkili olarak yavru kedi miyavlaması gibidir. Bu ağlayış sendromu tanımlar. Bu bebeklerin 1/3'ü 2 yaşından önce bu ağlayışlarını kaybederler. Cri du chat sendromunun diğer belirtileri ise şöyledir:[2]
- Yutma ve emmedeki zorluklar yüzünden beslenme problemleri,
- Düşük doğum ağırlığı ve zayıf büyüme,
- Bazı kavrama, heceleme ve motor gerilikler,
- Hiperaktivite, saldırganlık, huysuzluk nöbetleri ve tekrarlamalı hareketler gibi davranışsal problemler,
- Zamanla değişebilen alışılmamış yüz çehresi,
- Aşırı salya,
- Kabızlık,
- Gözler arası mesafenin geniş olması (hipertelorizm),
- Ense yapısının küçük olması,
- Yüksek ve dar damak yapısı,
- Parmaklar arasında kısmi perde.
Ek olarak, genel bulgular düşük doğum ağırlığı, hipotoni, mikrosefali, gelişme geriliği, dolgun yanaklı yuvarlak bir yüz, epikardiyum katları, düşük-eğilimli palpebral çatlaklar, strabismus, düz burun köprüsü, aşağı dönük ağız, retrognati, mikrognati, maloklüzyon, diş minelerinde defektler, diş sürmelerinde aksamalar, periodontal patolojiler izlenir.[3][4] Düşük kulaklar, kısa parmaklar, tekli avuç çizgisi ve kalple ilgili eksiklikler (örn; ventrikular septal defekt (VSD), atrial septaldefekt (ASD), PDA) vardır. Daha az sıklıkla görülen bulgular ise şöyledir: yarık dudak ve damak, timik displazi, hazım anomalisi, megakolon, kasık fıtığı, kalça çıkığı, kriptorsizm, hipospadias, az bulunan böbrek anomalileri (örn: at nalı böbrek, renal ektopi, hidronefroz), beşinci parmakta klinodaktili, talipes equinovarus, pes planus, ikinci ve üçüncü parmaklarda sindaktili, oligosindaktili ve aşırı-esneyebilen ek yerleri.
İleri çocukluk çağları ve ergenlikte zeka geriliği, mikrosefali, yüz simasının kabalaşması, çıkıntılar, derine yerleşik gözler, düşük burun kemeri, üst dişlerin sorunlu kapanışı ve belkemiğinde normal dışı yan kıvrım gibi bulgular da görülebilir.
Hasta dişi bireyler ergenliğe ulaştıklarında mensturasyonla birlikte ikincil eşey özellikleri geliştirirler. Genital bölge genellikle çift-uzantılı rahim şeklindeki bir rapor haricinde normaldir. Erkeklerde testisler genellikle küçüktür fakat spermatogenezin normal olduğu düşünümektedir. Cri du chat'den etkilenmiş insanlar doğurgandır ve üreyebilirler.
Genetiği
Cri du chat sendromu 5. kromozomun kısa kolunun kısmi bir delesyonu ile ilişkilidir. Vakaların %80'i ara sıra meydana gelen de novo delesyonlarla ilişkiliyken; %10-15'i genomun trizomik bir parçasınca eşlik edilen 5p monozomisinin bulunduğu ailesel düzenli bir translokasyonun düzensiz ayrılmasıyla ilişkilidir. Bu bireylerin fenotipleri genomdaki trizomik kısım fazlalığı nedeniyle 5p monozomisiyle sınırlananlardan daha belirgin olarak fark edilebilir.
Çoğu vaka 5p bölgesinin %30-60 uçsal delesyonlarını kapsar. %10'dan daha azı diğer nadir bulunan sitogenetik sapkınlıklarla (örn: interstitial delesyonlar, mozaizmler, ring kromozomlar ve de novo translokasyonlar) ile ilişkilidir. Bu durumlarda silinen 5. kromozomun kaynağı %80 ailesel kökenlidir.
Kayıpların çoğu kromozomun 5p15.2 - cri du chat kritik bölgesinde gerçekleşip, kedi miyavlaması haricindeki özelliklerden sorumluyken, 5p15.3 kedi-benzeri kritik bölgesinde gerçekleşen delesyon tipik ağlamadan sorumldur. Bu sonuçlar bu hastalığın etiyolojisinde bitişik olmayan iki bölgenin rol oynadığını ileri sürmektedir. Kritik bölgede haritalanmış Semaphorine F (SEMA5A) ve [delta catenin] (CTNND2) olarak isimlendiirlmiş iki genin Cri du Chat sendromlu hastaların beyinsel gelişimiyle muhtemelen ilişkilidir. Ayrıca telomeraz revers transkriptaz (hTERT) geninin bulunduğu 5p15.33 bölgesi bu sendromun fenotipini değiştirmeye katkıda bulunmakta gibi görünmektedir.
Delesyonun büyüklüğü her ne kadar değişse de, 5p15.3 bölgesindeki bir delesyon özgün ağlamadan, 5p15.2 kritik bölgesindeki delesyon ise diğer özelliklerden sorumludur. Sendromda %80 civarında olan ailesel kökenli delesyonlar de novo'dur.
Tedavi
Mevcut tedavi yöntemi yoktur. Sendromun neden olduğu sağlık problemleri (emme ve yutmada zorlanma, mide sorunları, kabızlık, şaşılık, böbrek ve kalp sorunları, solunumun geçici olarak durması, zayıf kas yapısı vs.) semptomatik yollarla tedavi edilebilir.
Ayrıca çocuklara erken dönemlerde konuşma ve davranış terapileri uygulanmalıdır. Sendromu taşıyan çocukların birçoğu eğitilebilir, kendilerine bakabilecek kadar sosyal gelişim gösterebilir.
Bu sendroma sahip bireylerin ebeveynlerinin genetik danışma alması ve çocuğa uygun eğitim planının yapılması aileye ve çocuğa daha kaliteli bir yaşamı sunar.
Kaynakça
- ^ Lejeune J, Lafourcade J, Berger R; ve diğerleri. (1963). "[3 Cases of partial deletion of the short arm of chromosome 5]". C. R. Hebd. Seances Acad. Sci. (Fransızca). Cilt 257. ss. 3098-102. PMID 14095841.
- ^ Cerruti Mainardi P, Perfumo C, Cali A, et al. Clinical and molecular characterization of 80 patients with 5p deletion: genotype-phenotype correlation. Journal of Medical Genetics, 38: 151-158, 2001
- ^ Hall C, Hallett K, Manton D. The association between Cri du chat syndrome and dental anomalies. Journal of Dentistry for Children (Chicago), 81(3):171-177, 2014
- ^ Corcuera-Flores JR, Casttellanos-Cosano L, Torres-Lagares D, et al. A systematic review of the oral and craniofacial manifestations of cri du chat syndrome. Clinical Anatomy, 29(5):555-560, 2016
Ayrıca bakınız
- Genetik hastalık
- Genetik tanı merkezi
Dış bağlantılar
- http://www.criduchat.asn.au 21 Mart 2019 tarihinde Wayback Machine sitesinde arşivlendi.
- http://www.fivepminus.org 1 Aralık 2020 tarihinde Wayback Machine sitesinde arşivlendi.
- http://www.criduchat.co.uk 22 Ocak 2009 tarihinde Wayback Machine sitesinde arşivlendi.
- http://criduchat.de 24 Kasım 2020 tarihinde Wayback Machine sitesinde arşivlendi. 21 yaşındaki bir Cri du chat sendromlu
- http://biyolojiegitim.yyu.edu.tr/mk/gh/cd.htm 8 Ağustos 2007 tarihinde Wayback Machine sitesinde arşivlendi.
| Sınıflandırma | |
|---|---|
| Dış kaynaklar |














