Kromozom 18, toplamda 22 çift olan otozomal insan kromozomlarından onsekizincisidir. İnsanlarda normalde bir çift halinde bulunur. 76 milyon baz çiftine ve toplam hücre DNA'sının %2,5'ine sahiptir. Kromozom 18, muhtemelen 300 ile 400 arasında gen içermektedir.

Down sendromu, trizomi 21 ya da mongolizm; genetik düzensizlik sonucu insanın 21. kromozom çiftinde fazladan bir kromozom bulunması durumu ve bunun sonucu olarak ortaya çıkan genetik bir bozukluktur. Down sendromu, bireyin 1 yaşından daha uzun süre yaşayabildiği tek otozomal trizomidir.
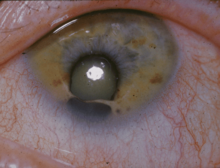
Turner sendromu, kromozom anomalisi sonucu ortaya çıkan ve kız çocuklarını etkileyen bir sendromdur; bir dişide eşey kromozomlarından birinin bulunmaması sonucu ortaya çıkar. Turner sendromluların fenotipi dişi olarak görülür fakat; eşey organları ve eşey hücreleri gelişmez. Kısır bireylerdir.
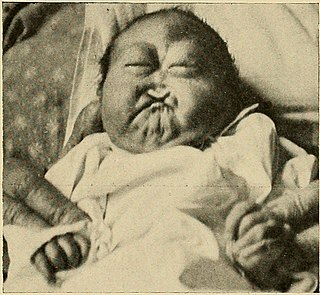
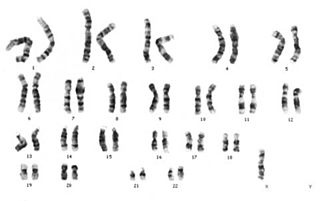
Patau sendromu ya da Trizomi 13, görülme sıklığı 10.000 canlı doğumda bir görülen bir kromozomal anomalidir.

Kedi miyavlaması sendromu, Kedi çığlığı sendromu veya tıptaki isimleriyle Cri du Chat sendromu ya da Cri-du-Chat sendromu, 5. kromozomun bir parçasının kaybıyla ilişkili nadir bulunan bir genetik düzensizliktir. Sendromun genetik tanımı 45,X(X/Y),-5p olarak gösterilir. Yani kişide 45 kromozomun bulunduğunu fakat 5. kromozomun kısa (petit) kolunun bir kısmının bulunmadığını ifade eder. Bu tip kromozom mutasyonlarında DNA'daki bazın ya da bazların yok olmasına delesyon adı verilir. Delesyondaki büyüklük bebeklerdeki fiziksel, psikomotor ve zihinsel gelişimlerini etkiler.
Hunter sendromu ya da Tip II mukopolisakkaridoz, iduronat-2-sülfataz (I2S) enziminin eksikliğinden ya da yokluğundan kaynaklanan lizozomal depo hastalığıdır. Doktor Charles A. Hunter (1873-1955) tarafından ilk kez 1917 yılında tanımlandığından, Hunter sendromu olarak adlandırılır.
Konjenital bozukluk olarak da bilinen doğum kusuru, nedeni ne olursa olsun doğumda mevcut olan anormal bir durumdur. Doğum kusurları fiziksel, zihinsel veya gelişimsel engelliliklerle sonuçlanabilir.

1p36 delesyon sendromu, orta ila şiddetli zeka geriliği, gecikmiş büyüme, hipotoni, nöbetler, sınırlı konuşma yeteneği, malformasyonlar, işitme ve görme bozukluğu ve farklı yüz özellikleri ile karakterize konjenital bir genetik bozukluktur. Belirtiler, kromozomal delesyonun tam yerine bağlı olarak değişebilir.
22q11.2 deletion sendromu, kromozom anomalisi kökenlidir; olguların bir bölümünün kalıtsal olduğu saptanmıştır. Çok sayıda fenotipi vardır.
LADD sendromu; lacrimo-auriculo-dento-digital sendrom; Levy-Hollister sendromu), ektodermal displazi bulguları da içeren, gözyaşı bezi,kulak, diş ve parmak bulgularının baskın olduğu, otosomal dominant yolla geçen kalıtsal bir sendromdur.
Mikrognati (micrognathism), altçenenin yetersiz gelişmesi (hipoplazi) niteliğinde bir anomalidir; 1. ve 2. faringeal arklara (brankial yarık) özgü malformasyonların çoğu çene-yüz bölgesindeki mezenkimal dokunun embriyolojik dönemdeki yetersizliğine bağlanmaktadır. Altçene küçüktür ve geridedir; bu olgu, çevredeki kasların hareketlerini sınırlarken bu kaslardan yararlanan dokuların gelişmeleri de kısıtlanır. Kraniyofasiyal anomalileri içeren sendromlarda çok sık görülen bulgulardan biridir. Sendroma-özgü olmayan olguların varlığı da bilinmektedir; örneğin; altçenesi, kafa ile göğüs kafesi arasında sıkışmış olan bir fetüste çene kemiklerinin gelişimi olması gereken düzeye ulaşamamaktadır. Gebelik sorunları, gebelerin alkol kullanması, gestasyonel diabet (gebelik diabeti) gibi çevresel faktörler etkili olabilmektedir (Möbius sendromu). Olguların bir bömümü, gebelerdeki rubella (kızamıkçık) infeksiyonu komplikasyonudur. Yenidoğanların bir bölümünde görülebilen hafif mikrognatiler, altçene gelişiminin tamalanmasıyla silinebilir. Mikrognati'de altçene gövdesi kadar çene eklemini oluşturan yapıların hipoplazisi de önemlidir. Mikrognatilerin bir bölümü kulak sistemi, üstçene ve damak gelişiminin de aksadığı olgularla birlikte görülür.
Möbius sendromu, maksillofasiyal, lokomotor ve nörolojik bulguların ön planda olduğu bir sendromdur. Olguların önemli bir bölümünü izole olgulardır; etyolojilerinde çevresel faktörlerin etkisi belirlenir. Otosomal dominant yolla aktarılan kalıtsal olgulara ancak birkaç ailede rastlanılmıştır. Oromandibular-limb hipogenezi grubu içinde yer alır. Yüzün bir yarısını etkileyen (unilateral) olgulara da rastlanmıştır.

Joubert sendromu, kalıtsal bir sendromdur; 36 fenotipinden 34'ü otosomal resesif yolla aktarılır, olguların bir bölümü Meckel sendromu ile çakışmaktadır. Joubert sendromunun tüm fenotiplerinde 3 temel bulgu vardır: santral sinir sistemi anomalileri, fizik gelişme geriliği, solunum sorunları. Serebellum (beyincik) vermisi ve beyin sapı hipoplazisi ile beyin radyolojisinde saptanan “azıdişi bulgusu ” santral sinir sistemi anomalilerinin başında gelir. Ataksi ve hipotoni sık görülür. Bazı hastalarda oksipital meningosel ya da meningomyelosel olabilir. Schinzel acrocallosal sendromu, önemli fenotiplerinden biridir.
Mikroftalmi sendromları, 18 fenotipi olan, etkilenen gen türüne göre farklı yollarla -otosomal dominant (AD), otosomal resesif (AR), X-kromozomu dominant (XLD), X-kromozomu resesif (XLR)- aktarılan kalıtsal patolojilerdir. Ortak bulgular yanı sıra farklı sistemlere özgü bulgularla da karşılaşılmaktadır. Ortak bulguların en büyük kümesi gözlerle ilgilidir.
Multipl konjenital anomaliler-hipotoni-epilepsi sendromu, 3 fenotipi olan kalıtsal bir sendromdur. Fenotip 1 (MCAHS1) ve fenotip 3 (MCAHS3) otosomal resesif, tip 2 (MCAHS2) ise x-kromozomu aracılığı ile resesif olarak aktarılır. Birçok enzimin çalışabilmesinde, kompleman sisteminin düzenlenmesinde ve hücrelerarası iletişimin sağlanmasında önemli katkıları olan, fosfogliserid yapısındaki glikosilfosfatidilinositol sentezindeki sorunlarından kökenlidir. Nörolojik belirtiler sendromun fenotiplerindeki temel bulguları oluştururlar; bunlara eklenen çok sayıdaki anomalilerin yeri, sayısı ve gücü farklıdır.

MVA sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. 3 fenotipi vardır; fenotip 1 en sık görülenidir. Anöploidi olarak niterlendirilen kromozom sayısı anomalisinden kökenlidir; kromozom eksikliği (monosomi) ya da fazlalığı (trisomi) vardır. Ancak, bazı hücrelerdeki kromozom sayısının normal olması nedeniyle “mozaisizm” grubu içinde yer alır.

Warkany sendromu, 2 fenotipi olan bir sendromdur. Fenotip 1 X-kromozomu aracılığıyla aktarılır. Fenotip 2’de ise kromozom 8 anomalisi nedeniyle spontan olarak ortaya çıkan izole olgulardır.
CHARGE sendromu ;, CHD7 genindeki spontan mutasyona bağlı izole olgulardır; az sayıda otosomal dominant yolla aktarılan kalıtsal örnekler de vardır. Gözlerdeki yapısal bozukluklar, kalp defektleri, üst solunum yollarındaki darlıklar, gelişme geriliği, genital hipoplaziler, kulak ve ekstremite anomalileri ön plandaki bulgulardır.

Potter sequence olgularında belirlenen önemli neden oligohidramnios bulgusudur.

Meckel-Gruber sendromu, çok sayıda genin etkilenmesinden kaynaklanan, bu nedenle 15 fenotipi olan, otosomal resesif yolla aktarılan, siliyopatiler grubundan, kalıtsal bir sendromdur. Kistik böbrekler, beyin anomalileri ve ensefalosel, polidaktili ve karaciğer anomalileri başlıca bulgulardır. En sık görülen fenotip Meckel sendromu 1, öteki fenotiplerdeki bulguların çok büyük bir bölümünü içerir. Bazı fenotipleri Joubert sendromuyla çakışan nitelikleri içermektedir.