Kavernom
| Kavernom | |
|---|---|
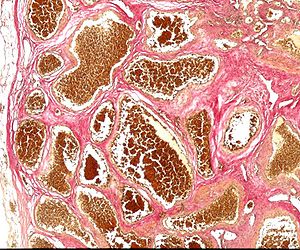 | |
| Kavernom patoloji kesiti | |
| Uzmanlık | Onkoloji, hematoloji, Kardiyoloji, Beyin ve sinir cerrahisi |
Serebral Kavernöz malformasyonlar, merkezi sinir sisteminin vasküler yapılarından kaynaklanan ve kavernöz hemanjiom olarak da isimlendirilen oluşumlardır. Hemanjiomların bir türü olarak değerlendirilebilir. Kavernomlar genişlemiş kan damarlarından, büyük vasküler kanallardan oluşur ve düzgün sınırlı, tek katlı endotelden oluşan, arasında normal beyin dokusunun bulunmadığı yapılardır. Serebral kavernöz malformasyonların çapı birkaç milimetreden birkaç santime kadar değişebilen büyüklüklerde olabilir. Sıklıkla beyinde görünür fakat başka organlarda da izlenebilir.[1]
Semptomlar
Klinik semptomlar, tekrarlayan baş ağrıları, bölgesel nörolojik kayıplar, hemorajik inme, nöbet olabilir. Çoğu hastada semptom izlenmez ve tesadüfi olarak başka sebeplerden dolayı çekilen beyin görüntülemelerinde tespit edilebilir.
Genetik
Ailesel olan Serebral kavernöz malformasyonlar (SKM) bilinen 3 gen lokusundan kaynaklanır. CCM1 geninin kodladığı ve ICAP1alpha (integrin cytoplasmic domain associated protein alpha)[2] proteinine bağlanan KRIT1[3][4] (krev interaction trapped 1) protein ve bir beta 1 integrin bağlantılı protein bulunmaktadır. CCM1 genindeki bazı mutasyonlar (q455x mutasyonu) Güneybatı Amerika'daki bazı hastalarda tespit edilmiştir.[5] Kuzey New Mexico'da bulunan bazı vakaların kökenleri eski İspanyol yerleşimcilere kadar tespit edilebilmiştir.[6]
CCM2 geni, malcaverin isimli bir protein kodlar ve bu proteinin Fosfotirozin bağlanma bölgesi bulunmaktadır.[7] CCM2 geninin görevi tam olarak bilinmemektedir. CCM2 proteininin hücrenin osmotik stres altında kaldığında p38 aktivasyonu için gerekli olan MAP kinase proteininin katlanmasını sağladığı düşünülmektedir.[8] Ayrıca Rac ve actin proteinlerine de bağlanmaktadır. CCM2 proteini ayrıca OSM (osmosensing scaffold for MEKK3) olarak da isimlendirilmektedir.
CCM3 geni en son tanımlanan gendir. CCM3 proteini, TF-1 isimli insan myeloid seri hücrelerinde apoptoz sırasında arttığı bilinen PDCD10 (programlı hücre ölümü 10) olarak bilinen protein olarak tanımlanmıştır.[9] CCM yolağındaki PDCD10 proteinin görevi tam olarak bilinmemektedir. Yakın zamanda PDCD10 proteinin CCM1 proteini olan KRIT1 ve CCM2 protini olan OSM ile kompleks oluşturduğu gösterildi. Tüm CCM proteinlerinin hücresel yolaklardaki ilişkileri ve fonksiyonları halen araştırılmaktadır.
Bazı kanıtlar SKM'lara sebep olabilecek 4. Bir gen olan CCM4 genini işaret etmektedir.[10] Serebral kavernöz malformasyonlu hastaların yaklaşık %70-80'inde bu gende mutasyon bildirilmiştir. Kalan %20-30 civarındaki hastaların ise halen tanımlanmamış başka gen mutasyonlarından kaynaklandığı düşünülmektedir. Yakın zamanda yapılan çalışmalarda SKM endotel hücrelerinde CDC42 gen delesyonu izlenmesi, bu genin CCM4 geni olabileceği şüphesi doğurmaktadır.[11]
Tanı
Tanı genellikle manyetik rezonans görüntüleme (MRG) ile konulmaktadır. Standart MRG sekanalarında gözden kaçabilen bu lezyon, özellikle MRG'nin gradient-echo ismi verilen özel bir sekansı ile net görüntülenmektedir. Standart T2 ağrılıklı görüntülerle mukayese edildiğinde FLAIR görüntülemelerde daha iyi izlenmektedir. FLAIR sekansları gradient-echo'dan farklıdır. FLAIR sekanslar T2 ağırlıklı sekanslara benzemekle birlikte FLAIR'de hareketli sıvı görüntüleri baskılanmaktadır. SKM'lar ayrıca tesadüfi olarak da beyin görüntülemelerinde tespit edilebilmektedir. Bunlara “insidentaloma” ismi verilmekte olup çoğunlukla semptom vermezler. Kanamaya sebep olduğu durumlarda yeni kanamanın ayrımının yapılması açısından bilgisayarlı tomografi daha özgüldür.
MRG'de bazen bu lezyonların ayırıcı tanısı yapmak güç olabilmektedir. Uzmanlar bu lezyonların ayırıcı tanısını yapmak için bazen anjiogram veya MRG-anjiogram (MRA) tetkikleri yapabilirler. SKM'lar yavaş akımlı vasküler yapılar oldukları için vasküler sistenin daha çok venöz yapılarına yakındırlar ve çoğunlukla anjiografik tetkiklerde tespit edilemezler. Eğer MRG'de izlenen lezyon anjiogramda da görüntülenebilirse öncelikli tanı arteriovenöz malformasyon lehinedir.
İnsidans
Genel populasyonda insidans %0,5 civarındadır ve klinik bulgular genellikle 2-30 yaşları arasında görülür.[12] Bu lezyonlar hemen her zaman konjenital'dir. Sporadik ve otozomal dominant kalıtım ile meydana gelebilirler.
Kaynakça
- ^ Batra, Batra (Aralık 2009). "Cavernous malformations: natural history, diagnosis and treatment". Nature Reviews Neurology. 5 (12). ss. 659-670. doi:10.1038/nrneurol.2009.177.
- ^ Zawistowski, Zawistowski (1 Şubat 2002). "KRIT1 association with the integrin-binding protein ICAP-1: a new direction in the elucidation of cerebral cavernous malformations (CCM1) pathogenesis". Human Molecular Genetics. 11 (4). ss. 389-396. doi:10.1093/hmg/11.4.389.
- ^ Couteulx, Couteulx (Ekim 1999). "Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas". Nature Genetics. 23 (2). ss. 189-193. doi:10.1038/13815.
- ^ Sahoo, Sahoo (1 Kasım 1999). "Mutations in the Gene Encoding KRIT1, a Krev-1/rap1a Binding Protein, Cause Cerebral Cavernous Malformations (CCM1)". Human Molecular Genetics. 8 (12). ss. 2325-2333. doi:10.1093/hmg/8.12.2325.
- ^ Petersen, Petersen (Şubat 2010). "Familial versus Sporadic Cavernous Malformations: Differences in Developmental Venous Anomaly Association and Lesion Phenotype". American Journal of Neuroradiology. 31 (2). ss. 377-382. doi:10.3174/ajnr.A1822.
- ^ Günel, Günel (11 Nisan 1996). "A Founder Mutation as a Cause of Cerebral Cavernous Malformation in Hispanic Americans". New England Journal of Medicine. 334 (15). ss. 946-951. doi:10.1056/NEJM199604113341503.
- ^ Liquori, Liquori (Aralık 2003). "Mutations in a Gene Encoding a Novel Protein Containing a Phosphotyrosine-Binding Domain Cause Type 2 Cerebral Cavernous Malformations". The American Journal of Human Genetics. 73 (6). ss. 1459-1464. doi:10.1086/380314.
- ^ Zawistowski, Zawistowski (1 Eylül 2005). "CCM1 and CCM2 protein interactions in cell signaling: implications for cerebral cavernous malformations pathogenesis". Human Molecular Genetics. 14 (17). ss. 2521-2531. doi:10.1093/hmg/ddi256.
- ^ Bergametti, Bergametti (Ocak 2005). "Mutations within the Programmed Cell Death 10 Gene Cause Cerebral Cavernous Malformations". The American Journal of Human Genetics. 76 (1). ss. 42-51. doi:10.1086/426952.
- ^ Liquori, Liquori (Ocak 2006). "Low frequency of PDCD10 mutations in a panel of CCM3 probands: potential for a fourth CCM locus". Human Mutation. 27 (1). ss. 118-118. doi:10.1002/humu.9389.
- ^ Castro, Castro (12 Nisan 2019). "CDC42 Deletion Elicits Cerebral Vascular Malformations via Increased MEKK3-Dependent KLF4 Expression". Circulation Research. 124 (8). ss. 1240-1252. doi:10.1161/CIRCRESAHA.118.314300.
- ^ Rigamonti, Rigamonti (11 Ağustos 1988). "Cerebral Cavernous Malformations". New England Journal of Medicine. 319 (6). ss. 343-347. doi:10.1056/NEJM198808113190605.