Kardiyomiyopati
| Kardiyomiyopati | |
|---|---|
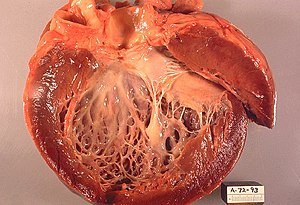 | |
| Kalınlaşma, genişleme ve kalbin içindeki beyazlığın artması olarak fark edilen subendokardiyal fibrozis gösteren açık sol ventrikül. | |
| Uzmanlık | Kardiyoloji |
| Belirtiler |
|
| Komplikasyon | |
| Tipler |
|
| Nedenleri |
|
| Tedavi | Türe ve semptomlara bağlıdır[5] |
| Sıklık | miyokardit ile 2,5 milyon (2015)[6] |
| Ölüm | miyokardit ile 354.000 (2015)[7] |
Kardiyomiyopati kalp kasının birincil hastalıkları grubudur.[1] Başlangıçta birkaç belirti olabilir veya hiç olmayabilir.[1] Hastalık kötüleştikçe, kalp yetmezliği başlangıcına bağlı olarak nefes darlığı, yorgunluk hissi ve bacaklarda şişme, düzensiz kalp atışı ve bayılma görülebilir.[1] Etkilenen kişilerde ani kalp ölümü riski vardır.[2]
2013 yılı itibarıyla kardiyomiyopatiler, "gözlemlenen fenotipe neden olmak için tek başına yeterli olan başka bir hastalığın yokluğunda morfolojik ve işlevsel olarak anormal miyokard ile nitelenen bozukluklar" olarak tanımlanır.[8][9]
Kardiyomiyopati türleri arasında hipertrofik kardiyomiyopati, genişlemiş kardiyomiyopati, kısıtlayıcı kardiyomiyopati, aritmojenik sağ ventrikül displazisi ve Takotsubo kardiyomiyopatisi (kırık kalp sendromu) bulunur.[3] Hipertrofik kardiyomiyopatide kalp kası büyür ve kalınlaşır.[3] Genişlemiş kardiyomiyopatide ventriküller büyür ve zayıflar.[3] Kısıtlayıcı kardiyomiyopatide ventrikül sertleşir.[3]
Çoğu durumda, neden belirlenemez.[4] Hipertrofik kardiyomiyopati genellikle kalıtsaldır, oysa genişlemiş kardiyomiyopati vakaların yaklaşık üçte birinde kalıtsaldır.[4] Genişlemiş kardiyomiyopati ayrıca alkol, ağır metaller, koroner arter hastalığı, kokain kullanımı ve viral enfeksiyonlardan da kaynaklanabilir.[4] Kısıtlayıcı kardiyomiyopati amiloidoz, hemokromatozis ve bazı kanser tedavilerinden kaynaklanabilir.[4] Kırık kalp sendromu aşırı duygusal veya fiziksel stresten kaynaklanır.[3]
Tedavi, kardiyomiyopati türüne ve semptomların şiddetine bağlıdır.[5] Tedaviler yaşam tarzı değişiklikleri, ilaçlar veya ameliyatı içerebilir.[5] Ameliyat, ventriküler destek cihazı veya kalp naklini içerebilir.[5]
2015 yılında kardiyomiyopati ve miyokardit 2,5 milyon kişiyi etkiledi.[6]
Hipertrofik kardiyomiyopati yaklaşık 500 kişiden 1'ini etkilerken, genişlemiş kardiyomiyopati 2.500 kişiden 1'ini etkilemektedir.[3][10] Bu hastalıklar 1990'daki 294.000'den 354.000 ölüme kadar yol açtı.[7][11]
Aritmojenik sağ ventrikül displazisi genç insanlarda daha yaygındır.[2]
Kardiyomiyopati ve ya kardiyomyopati, kalp kasını etkileyen hastalıklar için yapılan genel bir nitelemedir. Primer kardiyomiyopatiler ve Sekonder kardiyomiyopatiler olarak 2 ana grupta toplanırlar.
Belirtiler ve semptomlar

- Nefes darlığı veya özellikle fiziksel eforda nefes alma zorluğu,
- Yorgunluk
- Ayak bileklerinde, ayaklarda, bacaklarda, karında ve boyundaki damarlarda şişlik
- Baş dönmesi
- Fiziksel aktivite sırasında bayılma
- Aritmiler (anormal kalp atımları)
- Özellikle fiziksel efor veya ağır yemeklerden sonra göğüs ağrısı,
- Kalp üfürümleri (kalp atımlarıyla ilişkili alışılmadık sesler)
Nedenler
Kardiyomiyopatiler genetik (ailesel) veya genetik olmayan (edinilmiş) kökenli olabilir.[12]
Genetik kardiyomiyopatiler genellikle sarkomer veya sitoiskelet hastalıkları, nöromüsküler bozukluklar, doğuştan metabolizma hataları, kötü formasyon sendromları nedeniyle oluşur ve bazen tanımlanamaz.[13][14]
Genetik olmayan kardiyomiyopatilerin viral enfeksiyonlar, miyokardit ve diğerleri gibi kesin nedenleri olabilir.[15][16]
Kardiyomiyopatiler ya kalple sınırlıdır ya da genel sistemik bozukluğun parçasıdır ve her ikisi de sıklıkla kardiyovasküler ölüme veya ilerleyici kalp yetmezliğiyle ilişkili sakatlığa yol açar. Koroner arter hastalığı, yüksek tansiyon veya kalp kapakçıklarının anormallikleri gibi kalp kas işlev bozukluğuna neden olan diğer hastalıklar hariç tutulur.[17]
Genellikle altta yatan neden bilinmez kalır, ancak birçok durumda neden tanımlanabilir.[18] Örneğin alkolizm, ilaç zehirlenmesi ve bazı enfeksiyonlar (Hepatit C dahil) gibi genişlemiş kardiyomiyopatinin bir nedeni olarak tanımlanmıştır.[19][20][21] Tedavi edilmeyen çölyak hastalığı, zamanında teşhisle tamamen tersine dönebilen kardiyomiyopatilere neden olabilir.[22] Edinilmiş nedenlere ek olarak, moleküler biyoloji ve genetik çeşitli genetik nedenlerin tanınmasına yol açmıştır.[20][23]
Kardiyomiyopatinin 'hipertrofik', 'genişlemiş' veya 'kısıtlayıcı'[24] olarak daha klinik bir kategoriye sokulması, bazı durumların gelişimlerinin herhangi bir aşamasında bu üç kategoriden birden fazlasını karşılayabilmesi nedeniyle zorlaşmıştır.[25]
Amerikan Kalp Derneği (AHA) mevcut tanımı, kardiyomiyopatileri yalnızca kalbi etkileyen birincil ve vücudun diğer kısımlarını etkileyen hastalığın sonucu olan ikincil olarak ayırır. Bu kategoriler, yeni genetik ve moleküler biyoloji bilgilerini içeren alt gruplara da ayrılır.[26]
Mekanizma
Kardiyomiyopatilerin patofizyolojisi, moleküler tekniklerdeki ilerlemelerle hücresel düzeyde daha iyi anlaşılmaktadır. Mutant proteinler, kasılma aparatındaki (veya mekanosensitif komplekslerdeki) kardiyak fonksiyonu bozabilir. Kardiyomiyosit değişiklikleri ve hücresel düzeydeki kalıcı tepkileri, ani kardiyak ölüm ve diğer kardiyak sorunlarla ilişkili değişikliklere neden olur.[27]
Kardiyomiyopatiler genellikle bireysel olarak değişir. Farklı faktörler yetişkinlerde ve çocuklarda kardiyomiyopatilere neden olabilir. Örnek olarak yetişkinlerde dilate kardiyomiyopati, iskemik kardiyomiyopati, hipertansiyon, kapak hastalıkları ve genetikle ilişkilidir. Çocuklarda ise, X'e bağlı genetik bozukluk da dahil olmak üzere Becker kas distrofisi gibi nöromüsküler hastalıklar doğrudan kardiyomiyopatileri ile bağlantılıdır.[28]
Tanı

Kardiyomiyopatiyi belirlemek için yapılan tanı yöntemleri şunlardır:[29]
- Fiziksel muayene
- Aile geçmişi
- Kan tahlili
- EKG
- Ekokardiyogram
- Stres testi
- Genetik test
Sınıflandırma


Kardiyomiyopatiler farklı kriterler kullanılarak sınıflandırılabilir:[30]
- Birincil/içsel kardiyomiyopatiler[31]
- Doğuştan
- Hipertrofik kardiyomiyopati (HCM)
- Aritmojenik sağ ventrikül kardiyomiyopatisi (ARVC)
- Sol ventrikül sıkışmaması
- QT uzaması ve çok nadir görülen Kısa QT sendromu gibi İyon Kanalopatileri
- Katekolaminerjik polimorfik ventriküler taşikardi
- Karma
- Genişlemiş kardiyomiyopati (DCM)
- Kısıtlayıcı kardiyomiyopati (RCM)
- Brugada sendromu
- Edinilmiş
- Stres kardiyomiyopatisi
- Miyokardit, lenfositler ve monositler tarafından kısmen infiltrasyonundan kaynaklanan kalp dokusunun iltihaplanması ve yaralanması[32][33]
- Eozinofilik miyokardit, eozinofiller tarafından infiltrasyonundan kaynaklanan kalp dokusunun iltihaplanması ve yaralanması[32]
- İskemik kardiyomiyopati (iskemik kardiyomiyopatinin başka bir kardiyak problemin doğrudan sonucu olması nedeniyle sınıflandırmaya resmen dahil edilmemiştir)[31]
- Doğuştan
- İkincil/ekstrinsik kardiyomiyopatiler[31]
- Metabolik/depolama
- Fabry hastalığı
- Hemokromatozis
- Endomiyokardiyal
- Endomiyokardiyal fibrozis
- Hipereozinofilik sendrom
- Endokrin
- Kardiyofasiyal
- Nöromüsküler
- Kas distrofisi
- Friedreich ataksisi
- Diğer
- Obeziteyle ilişkili kardiyomiyopati[34]
- Metabolik/depolama
Primer kardiyomiyopatiler
Doğuştan gelen primer kardiyomiyopatiler
Hipertrofik kardiyomiyopati
Hipertrofik kardiyomiyopati (HCM), sol ventrikül kaslarının genellikle asimetrik kalınlaşmasıyla nitelenir.
Aritmojenik sağ ventriküler kardiyomiyopati


Daha önce aritmojenik sağ ventriküler displazi (ARVD) de denilen aritmojenik sağ ventriküler kardiyomiyopati (ARVCM), çoğunlukla doğuştan gelen bir hastalıktır.[35]
Veneto'da yaygınlık özellikle 1:2000 ila 1:5000 arasında yüksektir.[36] Ancak Almanya'da da ARVCM vakaları vardır. Hastalık ilerledikçe, sağ ventriküldeki giderek daha fazla kasın yerini yağ dokusu alır ve bu da sağ ventrikülün büyümesine neden olur. Kalbin pompalama fonksiyonundaki kısıtlamalar nadirdir. Rekabetçi sporlar gibi fiziksel stresin neden olduğu ani kalp ölümü (PHT), özellikle gençlerde daha yaygındır. Tanı ekokardiyografi, MR, EKG ve McKenna skoru kullanılarak konulabilir. Tedavi için kardiyoverter-defibrilatör implante edilebilir. Fiziksel efordan kaçınılmalıdır. Birçok ileri vakada kalp nakli son çaredir. ARVCM birçok vakada kalıtsal olduğundan etkilenen kişilerin aile üyelerinin muayene edilmesi önerilir. İtalya ve Amerika Birleşik Devletleri gibi bazı ülkelerde spor kulüplerinin tüm üyeleri profilaktik olarak taranmaktadır.
Aritmojenik sağ ventriküler kardiyomiyopatinin gelişmesinin bir nedeni desmozom proteinlerindeki mutasyonlar olabilir. Desmozomlar hücreler arasındaki hücre teması ARVCM durumunda özellikle miyokard için önemlidir. Örneğin Naxos hastalığında hücre yapışma proteini desmoplakini kodlayan DSP geni bir mutasyondan etkilenir. Genetik bozukluk, etkilenen hastalarda aritmojenik sağ ventriküler kardiyomiyopatiye yol açmaktadır.[37] Ara filament desmini kodlayan DES genindeki mutasyonlar da ARVCM'ye yol açabilmektedir.[38]
Hipertrofik kardiyomiyopati (HCM), görece sık görülen otosomal dominant genetik kalp hastalığıdır. Tabloya “Reggie Lewis hastalığı” denilmesinin nedeni, ABD'li basketbolcu Reggie Lewis'in maç sırasında bu hastalık nedeniyle ölmesidir. Olguların bir bölümünde klinik bulgular yüzeyseldir.[39][40][41][42]
Sol ventriküler hipertrabekülasyon
Özellikle kalp boşluğuna bağlanan kas lifleri (trabeküller) arasında derin boşluklar (sinüzoidler) bulunan sol ventrikülün ucundaki süngerimsi, şişmiş kaslarla birlikte görülen konjenital, nadir kalp kası hastalığıdır. Kalp kasının izole olmayan sıkışmasında (eşanlamlı: sıkışmayan kardiyomiyopati, sol ventriküler hipertrabekülasyon, süngerimsi miyokard), kalp kası erken embriyonik faz (süngerimsi miyokard) sırasında gevşek ağından daha çok sıkışmamıştır. İskelet kası hastalıklarında daha sık görülür ve ayrıca karmaşık siyanotik kalp kusurlarıyla birlikte görülür.
Tanı, kalp kateterizasyonu sırasında ekokardiyografi, MRI veya sol ventrikül anjiyografisi ile konur. Klinik gidişat belirsizdir. Şiddetli kalp yetmezliği, trombozlar, aritmiler ve ani kardiyak ölüm vakaları rapor edilmiştir. Z-disk, mitokondri ve tafazzin için G4.5 gen mutasyonlarının izole edildiği ailesel vakalar tanımlanmıştır.[43]
Glikojen depo hastalıkları
PRKAG2 ve bir glikojenoz tip II olan Danon arasında ayrım yapılır.
Hat kusurları
Lenègre hastalığı, His-Purkinje sisteminin birincil ilerleyici iletim kusurudur ve EKG'de QRS kompleksinin uzun duraklamalar, bradikardi ve senkopla birlikte genişlemesine yol açar.
Hasta sinüs düğümü sendromu fenotipik olarak iletim kusuruna benzer ve otozomal dominant şekilde ortaya çıkabilir.
Wolff-Parkinson-White sendromu (WPW) da nadiren ailelerde görülür.
Sekonder kardiyomiyopatiler
Sistemik (çokluorgan) hastalıkların komplikasyonu ya da bileşenidir. Kardiyomiyopatiye neden olan hastalıklar şunlardır:[39][40][41][42]
- Alkolizm
- Kimyasallar ve ilaçlar: Ağır metaller (kobalt, vd), emetine, kanser kemoterapisi
- Uyarıcı madde bağımlılığı: Kokain, metamfetamin
- Yüksek atım hacmi: Anemi, tirotoksikoz, gebelik
- İnfeksiyonlar (myokarditler): Virüsler (özellikle HIV), parazitler, protozoonlar (Chagas hastalığı)
- Endomyokardiyal patolojiler: Endomyokardiyal fibrozis, hipereozinofilik sendrom (Löeffler endokarditi)
- Hücre-dışı (intersellüler) madde birikmeleri: amiloidozis (primer, familyal otosomal dominant, senil, sekonder), Gaucher hastalığı (kalıtsal lipid depo hastalığı), mucopolysaccharidoses (kalıtsaldır)
- Hücre-içi (intrasellüler) madde birikmeleri (Kısıtlayıcı CM): Hemokromatozis, Fabry hastalığı (kalıtsal lizozomaldepo hastalığı), kalıtsal glikojen depo hastalığı (tip II, Pompe), Niemann-Pick hastalığı (kalıtsal lipid depo hastalığı)
- Elektrolit dengesizlikleri
- Endokrin hastalıklar: Diabetes mellitus, hipertiroidizm, hipotiroidizm, hiperparatiroidizm, feokromositoma, akromegali
- Nöromüsküler/nörolojik bozukluklar (tümü kalıtsaldır): Friedreich ataksisi, Duchenne-Becker müsküler distrofisi, Emery-Dreifuss müsküler distrofisi, Myotonik distrofi, Nörofibromatozis, Tuberoz skleroz
- Kardiyofasiyal kalıtsal sendromlar: Noonan sendromu, Lentiginosis
- Beslenme bozuklukları: Vitamin eksiklikleri (B ve C), selenyum eksikliği, karnitin eksikliği, Kwashiorkor, çinko eksikliği
- Granulomatöz yangılar (myokarditler): Sarkoidozis, dev hücreli myokardit
- Otoimmun hastalıklar (myokarditler): SLE, dermatomyositis, romatoid artrit, skleroderma, polyarteritis nodosa
- Kanser tedavisi: Anthracycline'ler, cyclophosphamide, radyoterapi
Klinik bulgular
Klinikte sinsi olarak gelişebilir (hastaların bazıları ekokardiyografik incelemelerde rastlantısal olarak belirlenir). Bulguların belirgin olduğu hastalarda EKG'de kalp hipertrofisi bulguları vardır.
Hipertrofik kardiyomiyopati hastalarında DNA analizi ile genetik köken araştırması yapıldığında kalıtım faktörü varlığı saptanır.
Gençlerde eforla gelen solunum güçlüğü (dispne), kalp ağrısı (angina pectoris) ve bayılma (senkop) önemli uyarılardandır. Bu nedenle, sporcu lisansı verilecek gençlerin kalp-damar ve solunum sistemlerinin ayrıntılı olarak incelenmesi gerekir. Hipertrofik kardiyomiyopati hastalarındaki önemli sorunlar şunlardır:[39][40][41][42]
- Atrial fibrilasyon ve mural trombus oluşması
- Mural trombuslardan kökenli embolizm
- Mitral kapakta infektif endokardit
- İnatçı konjestif kalp yetmezliği (tabloya kalp dilatasyonu da eklenebilir)
- Ansızın ölüm (sık görülen neden)
Patoloji
Kalp oldukça büyümüştür. Karıncıklararası bölmede (interventriküler septumda) düzensiz kalınlaşma (asimetrik septal hipertrofi) vardır. Mitral kapak düzeyindeki septum kalınlaşması sol karıncık (ventrikül) atımını olumsuz etkiler. Mikroskopta kalp kas liflerinde aşırı hipertrofi görülür. Kas dokusunun yerini yer yer bağ dokusunun aldığı adacıklar (fibrozis) vardır.[39][41]
Dilate kardiyomiyopati (DCM)
Dilate kardiyomiyopati, kalbin dört odacığının da hipertrofisi ve dilatasyonuyla nitelenir (dilatasyon, kalp kasının gevşemesi ve kalp boşluklarının ileri derecede genişlemesidir).[40][41][42] Ağırlığı 800 g'a kadar artan kalbin kasılmalarında yetersizlik ve bunun sonucunda sistolik işlev bozukluğu vardır. Sessizce gelişerek progressif konjestif kalp yetmezliğine dönüşebilir. İlk belirtilerden sonraki 5 yıl içinde hastaların %75'i kaybedilir.
Primer (idiopatik) DCM: Nedeni saptanamayan dilatasyon ve hipertrofi görülür. Olguların %30-40% olgu kalıtsaldır (otosomal dominant).
Sekonder DCM: Başka bir hastalığın komplikasyonu ya da parçasıdır. Dilate kardiyomiyopatilerin başlıca komplikasyonları:
- Kalbe gelen kan yüklenmesi
- Dolaşımda yavaşlama ve tromboz
- Akciğer ödemi
- Oksijensizlik
- Kalp yetmezliği
- Kardiyojen şok
- Ölüm
Kısıtlayıcı kardiyomiyopatiler (RCM)
Kısıtlayıcı kardiyomiyopatiler, kalp hareketlerini kısıtlayan kardiyomiyopatilerdir. Endokardın ve myokardın kalınlaşmış ve katılaşmıştır. Bu nedenle ventriküle kan girişi kısıtlanır.[41][42][44]
- Primer RCM: Löffler endokarditi, endokardiyal fibroelastozis gibi nedeni bilinmeyen olgulardır.
- Sekonder RCM: kalp hareketlerinin, myokardda yama biçiminde ya da yaygın bağ dokusunun artması (fibrozis) veya madde (amiloid) birikmesi gibi patolojiler sonucu kısıtlanmasıdır.
Kalp karıncık (ventrikül) duvarları normal ya da hafifçe kalınlaşmıştır. Ventriküllerde dilatasyonu yoktur. Myokard katılaşmıştır. Kulakçıklarda (atrium) dilatasyon sık görülür.
Özel kardiyomiyopati tipleri
Peripartum kardiyomiyopati (PPCM)
Doğumdan önceki ya da sonraki birkaç hafta içinde ortaya çıkan myokarditlerdir. En belirgin bulgu yüksek kan atım hacmıdır. Kronik hipertansiyon, beslenme bozuklukları, metabolik bozukluklar ve immunolojik kökenli tepkiler olası tetikleyicilerdir.[41][44]

Hipertansif kardiyomiyopati
Hipertansif kardiyomiyopati (hipertansif kalp hastalığı), hipertansiyonlu hastalarda görülen kalp büyümesi (hipertrofi) ve kalp hipetrofisinin neden olduğu olumsuzluklar kümesidir. Hipertansiyonun hızlandırdığı güçlü ateroskleroza bağlı kronik iskemik kalp hastalığı, kalp kas işlevinde aksamalara ve konjestif kalp yetmezliğine neden olur. İlerleyici böbrek yetmezliği (progresif renal yetmezlik), beyin kan dolaşımında aksamalar ve felç bu tablonun önemli bulgularıdır. İleri dönemlerde kalbin sağının etkilenmesiyle birlikte konjestif kalp yetmezliği belirir.[40][41][42]
Patoloji: Sol ventrikülde simetrik hipertrofi (ventrikül duvarı >2 cm) vardır, kalbin ağırlığı 500 g üzerindedir. Myokardda fibrozis (kronik iskemi bulguları) ve infarkt alanları görülür. Dekompanse olgularda dilatasyon saptanır. Akciğerler ödemlidir, alveol boşluklarında “kalp kusuru hücreleri” görülür. Böbreklerin dışyüzünde benekli görünüm izlenir (hipertansiyon bulgusu).
Tedavi
Tedavi, durumu daha iyi yönetmek için yaşam tarzı değişiklikleri önerisi içerebilir. Tedavi, kardiyomiyopati türüne ve hastalığın durumuna bağlıdır ancak yavaş kalp atım hızları için ilaç (muhafazakar tedavi) veya iatrojenik/implante edilmiş kalp pilleri, ölümcül kalp ritimlerine yatkın olanlar için defibrilatörler, şiddetli kalp yetmezliği için ventriküler destek cihazları (VAD'ler) veya ilaç veya mekanik kardiyoversiyonla ortadan kaldırılamayan tekrarlayan disritmiler için kateter ablasyonu içerebilir. Tedavinin amacı genellikle semptomların giderilmesidir ve bazı hastalar sonunda kalp nakline ihtiyaç duyabilir.[29]
Kaynakça
- ^ a b c d e "What Are the Signs and Symptoms of Cardiomyopathy?". NHLBI. 22 Haziran 2016. 15 Eylül 2016 tarihinde kaynağından arşivlendi. Erişim tarihi: 31 Ağustos 2016.
- ^ a b c "Who Is at Risk for Cardiomyopathy?". NHLBI. 22 Haziran 2016. 16 Ağustos 2016 tarihinde kaynağından arşivlendi. Erişim tarihi: 31 Ağustos 2016.
- ^ a b c d e f g h "Types of Cardiomyopathy". NHLBI. 22 Haziran 2016. 28 Temmuz 2016 tarihinde kaynağından arşivlendi. Erişim tarihi: 31 Ağustos 2016.
- ^ a b c d e "What Causes Cardiomyopathy?". NHLBI. 22 Haziran 2016. 15 Eylül 2016 tarihinde kaynağından arşivlendi. Erişim tarihi: 31 Ağustos 2016.
- ^ a b c d "How Is Cardiomyopathy Treated?". NHLBI. 22 Haziran 2016. 15 Eylül 2016 tarihinde kaynağından arşivlendi. Erişim tarihi: 31 Ağustos 2016.
- ^ a b GBD 2015 Disease and Injury Incidence and Prevalence Collaborators (8 Ekim 2016). "Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015". Lancet. 388 (10053). ss. 1545-1602. doi:10.1016/S0140-6736(16)31678-6. PMC 5055577 $2. PMID 27733282.
- ^ a b GBD 2015 Mortality and Causes of Death Collaborators (8 Ekim 2016). "Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015". Lancet. 388 (10053). ss. 1459-1544. doi:10.1016/s0140-6736(16)31012-1. PMC 5388903 $2. PMID 27733281.
- ^ Harrison's principles of internal medicine (21ci bas.). New York: McGraw Hill. 2022. s. 1954. ISBN 978-1-264-26850-4.
- ^ Arbustini, Eloisa; Narula, Navneet; Dec, G. William; Reddy, K. Srinath; Greenberg, Barry; Kushwaha, Sudhir; Marwick, Thomas; Pinney, Sean; Bellazzi, Riccardo; Favalli, Valentina; Kramer, Christopher; Roberts, Robert; Zoghbi, William A.; Bonow, Robert; Tavazzi, Luigi (3 Aralık 2013). "The MOGE(S) Classification for a Phenotype–Genotype Nomenclature of Cardiomyopathy". Journal of the American College of Cardiology (İngilizce). 62 (22). ss. 2046-2072. doi:10.1016/j.jacc.2013.08.1644. PMID 24263073. 19 Mayıs 2024 tarihinde kaynağından arşivlendi. Erişim tarihi: 12 Temmuz 2024.
- ^ Practical Cardiovascular Pathology. Lippincott Williams & Wilkins. 2010. s. 148. ISBN 978-1-60547-841-8. 14 Eylül 2016 tarihinde kaynağından arşivlendi.
- ^ GBD 2013 Mortality and Causes of Death Collaborators (17 Aralık 2014). "Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013". Lancet. 385 (9963). ss. 117-71. doi:10.1016/S0140-6736(14)61682-2. PMC 4340604 $2. PMID 25530442.
- ^ Bakalakos, Athanasios; Ritsatos, Konstantinos; Anastasakis, Aris (1 Eylül 2018). "Current perspectives on the diagnosis and management of dilated cardiomyopathy Beyond heart failure: a Cardiomyopathy Clinic Doctor's point of view". Hellenic Journal of Cardiology (İngilizce). 59 (5). ss. 254-261. doi:10.1016/j.hjc.2018.05.008. ISSN 1109-9666. PMID 29807197.
- ^ Rath, Anika; Weintraub, Robert (23 Temmuz 2021). "Overview of Cardiomyopathies in Childhood". Frontiers in Pediatrics. Cilt 9. s. 708732. doi:10.3389/fped.2021.708732. ISSN 2296-2360. PMC 8342800 $2. PMID 34368032.
- ^ Gorla, Sudheer; Raja, Kishore; Garg, Ashish; Barbouth, Deborah; Rusconi, Paolo (Aralık 2018). "Infantile Onset Hypertrophic Cardiomyopathy Secondary to PRKAG2 Gene Mutation is Associated with Poor Prognosis". Journal of Pediatric Genetics (İngilizce). 07 (4). ss. 180-184. doi:10.1055/s-0038-1657763. ISSN 2146-4596. PMC 6234042 $2. PMID 30430036.
- ^ Law, Michelle L.; Cohen, Houda; Martin, Ashley A.; Angulski, Addeli Bez Batti; Metzger, Joseph M. (February 2020). "Dysregulation of Calcium Handling in Duchenne Muscular Dystrophy-Associated Dilated Cardiomyopathy: Mechanisms and Experimental Therapeutic Strategies". Journal of Clinical Medicine (İngilizce). 9 (2). s. 520. doi:10.3390/jcm9020520. ISSN 2077-0383. PMC 7074327 $2. PMID 32075145.
- ^ Cimiotti, Diana; Budde, Heidi; Hassoun, Roua; Jaquet, Kornelia (8 Ocak 2021). "Genetic Restrictive Cardiomyopathy: Causes and Consequences—An Integrative Approach". International Journal of Molecular Sciences (İngilizce). 22 (2). s. 558. doi:10.3390/ijms22020558. ISSN 1422-0067. PMC 7827163 $2. PMID 33429969.
- ^ Lakdawala, NK; Stevenson, LW; Loscalzo, J (2015). "Chapter 287". Kasper, DL; Fauci, AS; Hauser, SL; Longo, DL; Jameson, JL; Loscalzo, J (Ed.). Harrison's Principles of Internal Medicine (19cu bas.). McGraw-Hill. s. 1553. ISBN 978-0-07-180215-4.
- ^ Lilly, Leonard S., (Ed.) (2011). Pathophysiology of heart disease: a collaborative project of medical students and faculty (5ci bas.). Baltimore, MD: Wolters Kluwer/Lippincott Williams & Wilkins. ISBN 978-1-60547-723-7. OCLC 649701807.
- ^ Adam A, Nicholson C, Owens L (2008). "Alcoholic dilated cardiomyopathy". Nurs Stand (Review). 22 (38). ss. 42-7. doi:10.7748/ns2008.05.22.38.42.c6565. PMID 18578120.
- ^ a b Westphal JG, Rigopoulos AG, Bakogiannis C, Ludwig SE, Mavrogeni S, Bigalke B ve diğerleri. (2017). "The MOGE(S) classification for cardiomyopathies: current status and future outlook". Heart Fail Rev (Review). 22 (6). ss. 743-752. doi:10.1007/s10741-017-9641-4. PMID 28721466.
- ^ Domont F, Cacoub P (2016). "Chronic hepatitis C virus infection, a new cardiovascular risk factor?". Liver Int (Review). 36 (5). ss. 621-7. doi:10.1111/liv.13064. PMID 26763484. 23 Eylül 2017 tarihinde kaynağından arşivlendi. Erişim tarihi: 12 Temmuz 2024.
- ^ Ciaccio EJ, Lewis SK, Biviano AB, Iyer V, Garan H, Green PH (2017). "Cardiovascular involvement in celiac disease". World J Cardiol (Review). 9 (8). ss. 652-666. doi:10.4330/wjc.v9.i8.652. PMC 5583538 $2. PMID 28932354.
- ^ Simpson S, Rutland P, Rutland CS (2017). "Genomic Insights into Cardiomyopathies: A Comparative Cross-Species Review". Vet Sci (Review). 4 (1). s. 19. doi:10.3390/vetsci4010019. PMC 5606618 $2. PMID 29056678.
- ^ Valentin Fuster; John Willis Hurst (2004). Hurst's the heart. McGraw-Hill Professional. s. 1884. ISBN 978-0-07-143225-2. 27 Mayıs 2013 tarihinde kaynağından arşivlendi. Erişim tarihi: 11 Kasım 2010.
- ^ Llamas-Esperón GA, Llamas-Delgado G (2022). "Hypertrophic cardiomyopathy. Proposal for a new classification". Arch Cardiol Mex. 92 (3). ss. 377-389. doi:10.24875/ACM.21000301. PMC 9262289 $2. PMID 35772124.
- ^ McCartan C, Maso R, Jayasinghe SR, Griffiths LR (2012). "Cardiomyopathy Classification: Ongoing Debate in the Genomics Era". Biochem Res Int. Cilt 2012. s. 796926. doi:10.1155/2012/796926. PMC 3423823 $2. PMID 22924131.
- ^ Harvey, Pamela A.; Leinwand, Leslie A. (8 Ağustos 2011). "Cellular mechanisms of cardiomyopathy". The Journal of Cell Biology. 194 (3). ss. 355-365. doi:10.1083/jcb.201101100. ISSN 0021-9525. PMC 3153638 $2. PMID 21825071.
- ^ Braunwald, Eugene (15 Eylül 2017). "Cardiomyopathies: An Overview". Circulation Research. 121 (7). ss. 711-721. doi:10.1161/CIRCRESAHA.117.311812. ISSN 1524-4571. PMID 28912178.
- ^ a b "What Are the Signs and Symptoms of Cardiomyopathy? - NHLBI, NIH". nhlbi.nih.gov. 28 Temmuz 2016 tarihinde kaynağından arşivlendi. Erişim tarihi: 25 Temmuz 2016.
- ^ Vinay, Kumar (2013). Robbins Basic Pathology. Elsevier. s. 396. ISBN 978-1-4377-1781-5.
- ^ a b c Maron, Barry J.; Towbin, Jeffrey A.; Thiene, Gaetano; Antzelevitch, Charles; Corrado, Domenico; Arnett, Donna; Moss, Arthur J.; Seidman, Christine E.; Young, James B. (11 Nisan 2006). "Contemporary Definitions and Classification of the Cardiomyopathies". Circulation (İngilizce). 113 (14). ss. 1807-1816. doi:10.1161/CIRCULATIONAHA.106.174287. ISSN 0009-7322. PMID 16567565. 20 Ağustos 2016 tarihinde kaynağından arşivlendi. Erişim tarihi: 1 Ağustos 2016.
- ^ a b Séguéla PE, Iriart X, Acar P, Montaudon M, Roudaut R, Thambo JB (2015). "Eosinophilic cardiac disease: Molecular, clinical and imaging aspects". Archives of Cardiovascular Diseases. 108 (4). ss. 258-68. doi:10.1016/j.acvd.2015.01.006. PMID 25858537.
- ^ Rose NR (2016). "Viral myocarditis". Current Opinion in Rheumatology. 28 (4). ss. 383-9. doi:10.1097/BOR.0000000000000303. PMC 4948180 $2. PMID 27166925.
- ^ Lipshultz, Steven E.; Messiah, Sarah E.; Miller, Tracie L. (5 Nisan 2012). Pediatric Metabolic Syndrome: Comprehensive Clinical Review and Related Health Issues. Springer Science & Business Media. s. 200. ISBN 978-1-4471-2365-1. 29 Mayıs 2016 tarihinde kaynağından arşivlendi.
- ^ G. Thiene, D. Corrado, C. Basso: Arrhythmogenic right ventricular cardiomyopathy/dysplasia. In: Orphanet Journal of Rare Diseases, 2007, Band 2, S. 45, ISSN 1750-1172. doi:10.1186/1750-1172-2-45. PMID 18001465. PMC 2222049. (Review).
- ^ A. Nava, G. Thiene u. a.: Familial occurrence of right ventricular dysplasia: a study involving nine families. In: Journal of the American College of Cardiology, November 1988, Band 12, Nummer 5, S. 1222–1228, ISSN 0735-1097. PMID 3170963.
- ^ N. Protonotarios, A. Tsatsopoulou: Naxos disease: cardiocutaneous syndrome due to cell adhesion defect. In: Orphanet Journal of Rare Diseases, 2006, Band 1, S. 4; ISSN 1750-1172. doi:10.1186/1750-1172-1-4. PMID 16722579. PMC 1435994. (Review-Artikel im Open Access).
- ^ B. Klauke, S. Kossmann u. a.: De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. In: Human molecular genetics, Dezember 2010, Band 19, Nummer 23, S. 4595–4607, ISSN 1460-2083. doi:10.1093/hmg/ddq387. PMID 20829228.
- ^ a b c d Ponikowski P, Voors AA, Anker SD, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. European Journal of Heart Failure (Review), 18 (8): 891–975, 2016
- ^ a b c d e Silnernagl S, Lang F. Color Atlas of Pathophysiology. Thieme, Stuttgart-New York, 2000
- ^ a b c d e f g h Kumar V, Abbas AK, Aster JC. Robbins and Cotran Pathologic Basis of Disease. 9th edt., Elsevier Saunders, Philadelphia, 2015
- ^ a b c d e f Westphal JG, Rigopoulos AG, Bakogiannis C, et al. The MOGE(S) classification for cardiomyopathies: current status and future outlook. Heart Fail Rev (Review), 22 (6): 743–752, 2017
- ^ Engberding, C. Stöllberger, P. Ong, T. Yelbuz, B. Gerecke, G. Breithardt: Isolierte Noncompaction-Kardiomyopathie. 16 Eylül 2011 tarihinde Wayback Machine sitesinde arşivlendi. In: Dtsch Arztebl Int., 2010; 107(12), S. 206–213.
- ^ a b Yücesoy G, Bodur H, Özkan S, Tekin A. HELLP Sendromlu ve Ağır Preeklamptik İki Farklı Olguda Peripartum Kardiyomyopati. Türkiye Klinikleri J Gynecol Obst., 15(6):328-332, 2005