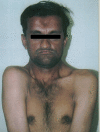
Cleidocranial dysostosis, kemik malformasyonları içeren kalıtsal bir sendromdur; ebeveynlerden birinden otosomal dominant yolla gelir. Genetik incelemeler, kemik gelişimiyle ilgili RUNX2 genindeki bir sorunu göstermektedir. 3 tipi vardır; dişlerin etkilenmesi bunlardan yalnızca birinde saptanır. İskelet sistemi etkilenmesinin en tipik bulgusu köprücük kemiklerinde (klavikula;clavicula) görülür. Köprücük kemikleri ya hiç oluşmamıştır (agenezis) ya da gelişmesi yetersizdir (hipoplazi); bu nedenle, hastalar, omuzlarını orta çizgi üzerinde birleştirebilirler. Kafatasındaki bıngıldaklar ve pelvisteki simfizi uzun süre kapanamaz.
Kraniyoektodermal displazi (Cranioectodermal dysplasia), ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur. 4 fenotipi vardır:
- Cranioectodermal dysplasia 1 (Sensenbrenner sendromu; Levin sendromu; arthrodentoosseous dysplasia)
- Cranioectodermal dysplasia 2
- Cranioectodermal dysplasia 3
- Cranioectodermal dysplasia 4
OPD sendromu (otopalatodigital sendrom), X-kromozomu aracılığıyla dominant (XLD) yolla geçen kalıtsal bir sendromdur (Gorlin-Cohen sendromu hariç). Kız çocukları daha hafif etkilenir. Tümü benzer bulgular içeren 5 tipi vardır:
- Otopalatodigital sendrom tip I (OPD tip1)
- Otopalatodigital sendrom tip II (OPD tip2)
- Gorlin-Cohen sendromu (ilgili sayfaya gidiniz)
- Melnick-Needles sendromu (tip 1 ile allelik bağlantısı vardır; ilgili sayfaya gidiniz)
- Terminal osseöz displazi ve pigmentli deri defektleri (çok enderdir, ayrıntı verilmemiştir).
Gorlin-Cohen sendromu , ektodermal displazi bulgularını da içerebilen bir OPD sendromu tipidir. 2 fenotipi vardır:
- Frontometaphyseal dysplasia 1
- Frontometaphyseal dysplasia 2
Melnick-Needles sendromu , X-kromozomu aracılığıyla dominant (XLD) yolla geçen kalıtsal bir sendromdur. Kız çocukları daha hafif etkilenir. Erkek çocuklarında güçlü OPD tip 2 bulguları saptanır; çoğu ölü doğar. Gelişme geriliği olabilir. Başlıca bulgular şunlardır:

Marshall sendromu, ektodermal displazi bulguları da içerebilen, otosomal dominant yolla geçen kalıtsal bir sendromdur.
Kraniyosinostoz, kraniyosinostozis (craniosynostosis), kraniyofasiyal malformasyonların ve maksillofasiyal sendromların önemli bir bölümünde etkileri görülebilen konjenital bir patolojidir. Bu olgudaki temel bulgu kafatası eklemlerinin erken kapanmasıdır; etkilediği anatomik bölgelerde ortaya çıkan malformasyonlar, hangi suturaların ne düzeyde kapanmış olmasıyla orantılıdır. Malformasyonlar genellikle etkilenen eklemin dikey yönünde belirgindir.
Even-Plus sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Genel gelişme geriliği vardır. Yenidoğanda, deri malformasyonu saptanır. Brakisefalik kafa biçimindeki kafatasında saçlar seyrektir. Boyun kısa, kaş çıkıntıları belirgindir. Kaşlar birbirine birleşmiş olabilir. Kulak deliği yoktur ya da dardır (microtia). Burun hipoplaziktir, ucunda yarık vardır. Yüz orta bölüm hipoplazisi nedeniyle üstçene küçüktür (mikrognati), damak çukuru belirgindir. Üst çenede tek orta kesici (santral) diş vardır, lateral dişler eksiktir (hipodonti). Kalpte konjenital anomaliler saptanır. Anüste darlık olabilir. Üriner sistemde, böbrek hipoplazisi ve infeksiyonlar olabilir. İskelet sisteminde, omurga yarıkları ve koksiks agenezi saptanır; uzun kemiklerde malformasyonlar vardır. Atopik dermatit olabilir. Corpus callosum agenezi, beyinde saptanan en önemli anomalidir.
Femoral-facial sendrom, izole sendromdur; diabetik annelerin bebeklerinde görece sık rastlanır. otosomal dominant yolla aktatılmış az sayıda olgu bildirilmiştir.
Frontonazal displazi, otosomal resesif yolla aktarılan, alın ve burun bölgelerinin malformasyonlarıyla karakterize kalıtsal bir sendromdur; ALX3 genindeki mutasyonun sonucudur. En önemli bulgu, frontal kemikte ve yüz kemiklerinde orta çizgi yarıklarının bulunmasıdır ile bunun sonucunda ortaya çıkan nazofrontal ensefalosel’dir. Alın derisinde lipoma olabilir. Mikroftalmi saptanır, gözler birbirinden aşırı uzaktadır (hipertelorizm); ptozis, kapak bileşkelerinde anomaliler, katarakt ve epibulbar dermoidler belirlenir. Kulak kepçeleri aşağıdadır, işitme sorunları vardır. Burun kökü aşırı yayvandır; bu alanda saptanan malformasyonların en önemlisi yarıktır. Üstçenede, paranazal sinüslerde ve frontal sinüslerde hipoplazi izlenir. Kalpte Fallot tetralojisi saptanabilir. Pektoral kaslar hipoplazi ya da agenez olabilir. El parmakları kısa ve kıvrıktır. Beyinde Corpus callosum anomalileri saptanır, zeka geriliği vardır. Akromelik frontonazal disostoz fenotipindeki bulgular daha azdır.

Goltz sendromu, X kromozomu aracılığıyla dominant (XLD) olarak aktarılan kalıtsal bir sendromdur; erkek fetüslerin çok büyük bölümü intrauterin evrede (rahimde) öldüğü için hastaların çoğu kız bebeklerdir.
Pallister-Hall sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Hipotalamus'ta hamartoblastoma olarak nitelendirilen oluşumun yanı sıra çok sayıda endokrin sistem anomalileri, açılmamış anüs ve polidaktili bulguları ön plandadır. Hipofiz agenezi/hipoplazisi kökenli hipopituitarizm, adrenal gland hipoplazisi bulguları, hipotiroidizm, endokrin sistem anomalilerinin başlıcalarıdır.
Raine sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Yeni doğan ve bebek ölümleri sıktır; çoğu doğumdan sonraki haftalar içinde kaybedilen bebeklerde, kemiklerde yaygın yoğunlaşmalar (skleroz) saptanır. Özellikle kafa tabanı ve yüz kemiklerindeki skleroz oldukça belirgindir. Ayrıca, yoğun bir periostal kemik yapımı nedeniyle, uzun kemiklerin çevresinde sklerotik bir kılıf oluşur. Mikrosefali ve alın bombesi oldukça yüksek olan brakisefali (turribrakisefali) saptanır. Bıngıldaklar (fontaneller) tam kapanmamıştır. Kafatasındaki kemik yoğunlaşması (osteoskleroz) kraniyofasiyal displazi bulgularının oluşmasına yol açar. Ekzoftalminin yanı sıra hipertelorizm de olabilir. Kulaklar arkaya dönük ve biçimsizdir. Yüz orta bölümü hipoplazisi nedeniyle üstçene küçüktür (mikrognati), basık ve silik bir burun yapısı izlenir; bu yapı balık yüzü izlenimi verir. Altçene önde gibidir (prognatizm). Ağız açıklığı dardır. Derinliği fazla olan bir yarık damak varlığı beslenme bozukluklarına neden olur. Doğumda dişler olabilir, zamanında süren dişlerse küçüktür (mikrodonti). Hastaların büyük bölümünde dişeti büyümeleri vardır. Dil oldukça büyüktür (makroglossi).

MVA sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. 3 fenotipi vardır; fenotip 1 en sık görülenidir. Anöploidi olarak niterlendirilen kromozom sayısı anomalisinden kökenlidir; kromozom eksikliği (monosomi) ya da fazlalığı (trisomi) vardır. Ancak, bazı hücrelerdeki kromozom sayısının normal olması nedeniyle “mozaisizm” grubu içinde yer alır.

Sipondilokarpotarsal sinoztozis sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. İskelet sisteminin tümünü ilgilendiren anomalilerin yanı sıra, özellikle gövde ve omurga sisteminin gelişmesindeki gerilik nedeniyle kısa boy ve orantısız bir vücut yapısı vardır. Parmak eklemlerinde kaynaşmalar görülür. Skapula, sternum ve kaburga anomalileri izlenir; çok sayıdaki vertebta anomalileri nedeniyle skolyoz ve lordoz saptanır. Toraksı olumsuz biçimde etkileyen çevre kemik anomalilerinin yol açtığı restriktif akciğer hastalığına bağlı solunum sorunları yaşarlar. Uzun kemiklerin epifizleri gelişememiştir ve eklemleri gevşektir. Dirsek eklemi hareketleri çok kısıtlıdır.

Konjenital spondiloepifizeal displazi , otosomal resesif yolla aktarılan, kondrodisplazi grubundan, iskelet sistemi malformasyonlarının yoğun olduğu kalıtsal bir sendromdur. Doğumdan önce belirlenen cücelik düzeyinde gelişme geriliği vardır. Cüceliğin nedeni gövde kısalığı, uzun kemiklerin epifizlerindeki malformasyonlar ve yassılaşmış omurgalardır. Boyun kısadır.

Toriello-Carey sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Doğum ölçümleri normaldir, ancak zamanla gelişme geriliği bulguları belirir.

Mandibulofacial dysostosis sendromu, yüz bulgularının ön planda olduğu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. 1. ve 2. farengeal arklardaki gelişme bozukluğunun sonucudur. "Mikrosefali içeren tipi " ve "Alopesi" içeren tipi önemlidir. “Bauru tipi” çok enderdir. “Ptozis" içeren tip ise kaynaklarda bildirilmiş tek olgu olarak görülmektedir. Treacher Collins sendromu ile yakınlığı tartışılmaktadır.

Asfiksiyan torasik displazi sendromu, otosomal resesif yolla aktarılan, 22 fenotipi olan kalıtsal bir sendromdur. İlk tanımlanan fenotip 1, Jeune sendromu olarak bilinmemektedir. Fenotiplerden ikisi Ellis-van Creveld sendromu kapsamındadır. Saldino-Noonan sendromu, Majewski sendromu, Mainzer-Saldino sendromu ve Beemer-Langer sendromu görece sık rastlanan fenotiplerdir.

Akromelik frontonazal disostoz, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Frontonazal displazilerin ender görülen tiplerinden biridir. Kafatası, çene ve yüz bulgularının yanı sıra görülebilen beyin ve iskelet sistemi bulguları önemlidir.
