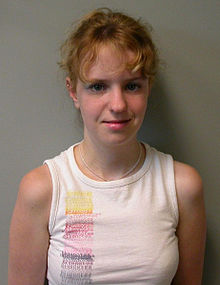Williams sendromu , 7. kromozomun uzun kolunda 26 genin silinmesiyle ortaya çıkan; ektodermal displazi bulguları da içeren, otosomal dominant geçen kalıtsal bir sendromdur. Genel gelişme geriliği izlenir, hastaların çoğu zayıftır. Kafatasındaki gelişme duraklamasının sonucu olarak oldukça geniş bir alın vardır. Yabancılara kolay güvenme, geç gelişen dil becerileri, kalp rahatsızlığı, geç gelişen koordinasyon-denge becerisi gibi sonuçlar doğuran nörolojik bozukluktur. Algılama (kognitif) sorunları vardır, psikiyatrik bulgularla karşılaşılabilir. Ses telleri felci nedeniyle ses kabadır. Hasta aktiftir ve mutlu bir görünüm ile aşırı dostça davranış sergiler. Uyku sorunları ve zeka geriliği olabilir.

Ellis-van Creveld sendromu, ektodermal displazi bulguları içeren, otosomal resesif geçen kalıtsal bir sendromdur. Asfiksiyan torasik displazi sendromu'nun 22 fenotipinden 2'si Ellis-van Creveld sendromu olarak değerlendirilir.
KID sendromu (keratitis-ichthyosis-deafness), ektodermal displazi ve sağırlık bulgularını bulgularını içeren, otosomal dominant yolla aktarılan kalıtsal bir sendromdur.

Coffin-Siris sendromu, ektodermal displazi bulgularını da içeren, otosomal dominant geçen kalıtsal bir sendromdur. 8 fenotipi vardır. Çene-yüz bulguları fenotiplerden 3'ünde belirgindir. Hastalarda genel gelişme geriliği vardır. Kafatası küçüktür (mikrosefali).
Costello sendromu, ektodermal displazi bulguları da içeren, otosomal dominant geçen kalıtsal bir sendromdur. Deri, yüz ve iskelet sistemi bulguları ön plandadır. Spontan gen mutasyonuna bağlı az sayıda olgu vardır. Doğumda iri bebek (makrosomi) olsa da, ileriki aylarda belirgin bir gelişme geriliği saptanır. Kısa bir boyun ve büyük bir kafatası (makrosefali) vardır. İnce-kıvırcık-seyrek erken dökülen saçlar erken yaşlanma (progeria) görümü oluşturur.
GAPO sendromu, ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur. Hastalarda genel bir gelişme geriliği vardır. Alın bombesi yüksektir. Saçlar giderek dökülür. Optik atrofi ve glokomun neden olduğu görme sorunları ile göz titremeleri (nistagmus) saptanır.

Marshall sendromu, ektodermal displazi bulguları da içerebilen, otosomal dominant yolla geçen kalıtsal bir sendromdur.

Agnati-Otosefali kompleksi, öncelikle mikrognatiye neden olan malformasyonlardan biridir; altçene yokluğu anlamına gelse de, altçenenin tümü değil ancak bir bölümünün eksikliği ya da oldukça aşırı bir hipoplazisi söz konudur. Ender görülen bu kompleksin kalıtsal olabileceği gibi, gen mutasyonlarının ve teratojenlerin etkisini gösteren olgular da bildirilmiştir.
Möbius sendromu, maksillofasiyal, lokomotor ve nörolojik bulguların ön planda olduğu bir sendromdur. Olguların önemli bir bölümünü izole olgulardır; etyolojilerinde çevresel faktörlerin etkisi belirlenir. Otosomal dominant yolla aktarılan kalıtsal olgulara ancak birkaç ailede rastlanılmıştır. Oromandibular-limb hipogenezi grubu içinde yer alır. Yüzün bir yarısını etkileyen (unilateral) olgulara da rastlanmıştır.

Bosma arini mikroftalmi sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; göz ve burun bulguları ön plandadır.

Emanuel sendromu, t(11;22)(q23;q11.2) ayrımı bozukluğu olarak tanımlanan bir kromozom anomalisi sendromudur.

Kabuki sendromu, iki fenotipi olan bir sendromdur: Kabuki sendromu 1 ve Kabuki sendromu 2 (Kabuk2). Kabuk1, otosomal dominant yolla aktarılan kalıtsal ya da izole olgular biçiminde görülür; Kabuk2 ise, X-kromozomu aracılığıyla dominant yolla (XLD) aktarılır. Her iki fenotipini çok sayıda ortak bulguları vardır; özellikle yüz, iskelet sistemi, gelişme geriliği, parmakizleriyle ilgili bulgular ve zeka geriliği önemlidir.
Mikroftalmi sendromları, 18 fenotipi olan, etkilenen gen türüne göre farklı yollarla -otosomal dominant (AD), otosomal resesif (AR), X-kromozomu dominant (XLD), X-kromozomu resesif (XLR)- aktarılan kalıtsal patolojilerdir. Ortak bulgular yanı sıra farklı sistemlere özgü bulgularla da karşılaşılmaktadır. Ortak bulguların en büyük kümesi gözlerle ilgilidir.

Cantu sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur.

3MC sendromu, otosomal resesif yolla aktarılan ya da spontan gen mutasyonu sonucu izole olgu olarak ortaya çıkan bir sendromdur. 3 fenotipi vardır. Önceleri Malpuech, Carnevale, Mingarelli ve Michels sendromları olarak bilinen sendromlar bu grup 3MC sendromu kapsamına alınmıştır. Göz kapakları malformasyonları, hipertelorizm, belirgin kaş çıkıntıları, kraniyofasiyal yarıklar, işitme sorunları ve algılamada güçlükler 3MC sendromunun fenotiplerinde çoğunlukla saptanan ortak bulgulardır.

Bohring-Opitz sendromu, otosomal dominant yolla aktarılan, gelişme ve zeka geriliği bulgularının ön planda olduğu, hastaların çoğunun çocukluk yaşlarında kaybedildiği bir sendromdur. C sendromu'nun fenotipi olarak benimsenir.

Hartsfield sendromu, otosomal dominant yolla aktarılan, ender görülen, kalıtsal bir sendromdur. El ve ayak parmaklarının konjenital eksikliği (ektrodaktili), yarık dudak ve yarık damak ile kafatası malformasyonu (holoprosensefali) üçlüsü temel bulgulardır.

Asfiksiyan torasik displazi sendromu, otosomal resesif yolla aktarılan, 22 fenotipi olan kalıtsal bir sendromdur. İlk tanımlanan fenotip 1, Jeune sendromu olarak bilinmemektedir. Fenotiplerden ikisi Ellis-van Creveld sendromu kapsamındadır. Saldino-Noonan sendromu, Majewski sendromu, Mainzer-Saldino sendromu ve Beemer-Langer sendromu görece sık rastlanan fenotiplerdir.

Vici sendromu, psikomotor gelişme geriliğinin yanı sıra göz, dolaşım sistemi ve bağışıklık sistemindeki defektlerle karakterize, otosomal resesif yolla aktarılan kalıtsal bir sendromdur.

Wolf-Hirschhorn sendromu, genel gelişme geriliği, kafa ve yüz malformasyonları ve epileptiform atakların görüldüğü, “4. kromozomun kısa kolunun distal kısmında delesyon (4p-)” olarak bilinen kromozom anomalisi sonucu ortaya çıkan izole olgulardır. Kız hastalar görece sıktır; 1/3’ü iki yaşına ulaşamadan kaybedilir.