
Şizofreni, benzer belirtilere sahip birtakım ruhsal hastalıklardır.
Ailevi Akdeniz ateşi veya eskiden yabancı literatürde Periyodik hastalık veya Ermeni hastalığı, sıklıkla Ermeni, Yahudi, Türk, Orta Doğu Arap toplumlarında ve Japonlarda görülen irsi ateşli hastalıktır. Ailevi Akdeniz Ateşi, tekrarlayan ateş, karın ağrısı, göğüs ağrısı ve eklem ağrısı nöbetleri yapan bir hastalıktır. Nöbetler genellikle 24-48 saat sürer. Hastalarda nöbetler dışında hiçbir belirti yoktur.

Parkinson hastalığı (PH) veya kısaca Parkinson, başlıca merkezî sinir sisteminin etkilendiği, uzun süreli bir nörodejeneratif hastalıktır ve hem motor hem de motor olmayan sistemleri etkiler. Semptomlar genellikle yavaş yavaş ortaya çıkar ve hastalık ilerledikçe motor olmayan semptomlar daha yaygın hale gelir.

Epilepsi ya da sara, beyin içinde bulunan sinir hücrelerinin olağan dışı bir elektro-kimyasal boşalma yapması sonucu ortaya çıkan sinirsel bozukluktur. Beynin normal faaliyetlerini sürdürmesini sağlayan elektriğin, aşırı ve kontrolsüz yayılımı sonucu oluşur. Sıklıkla geçici bilinç kaybına neden olur. Epilepsi nöbetleri farklı şekillerde ortaya çıkar. Bazı nöbetlerden önce korku hissi gibi olağan dışı algılamalar ortaya çıkarken, bazı nöbetlerde kişinin ağzı köpürebilir veya kişi yere düşebilir. Bu da kemik kırılması dâhil bazı fiziksel yaralanmalara sebep olabilir.
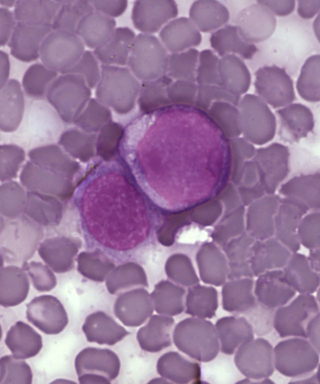
Lösemi, kan hücrelerinin özellikle de akyuvarların normalin üzerinde çoğalması ile kendini gösteren bir kanser türüdür.

Lyme hastalığı veya Borreliosis, genelde Ixodes ricinus (sakırga) türü kenelerin ısırması ile insana geçen Borrelia burgdorferi adlı ve benzer bakterinin yol açtığı bir hastalıktır.
Amyotrofik lateral skleroz (ALS), aynı zamanda motor nöron hastalığı olarak da anılan, merkezî sinir sisteminde, omurilik ve beyin sapı adı verilen bölgede motor sinir hücrelerinin kaybından ileri gelen bir hastalıktır. Bu hücrelerin kaybı kaslarda güçsüzlük ve erimeye (atrofi) yol açar. Ayrıca erken ya da geç hareketin birinci nöronu da hastalanır. Zihinsel fonksiyonlar ve bellek ise bozulmaz.
Hashimoto tiroiditi vücudun kendi koruma sisteminin tiroid hücrelerine saldırdığı bir bağışıklık sistemi sorunudur: Kandaki anti-TSH reseptör antikorlarının neden olduğu otoimmun bir hastalıktır.

Fenilketonüri, Fenilketonuria ya da PKU; otozomal çekinik kalıtım gösteren metabolik bir hastalıktır. Hasta kişiler fenilalanin amino asitini tirozin amino asitine çeviremezler. İki amino asit arasındaki tek fark tirozinde bulunan hidroksil grubudur (-OH). Fenilalanini tirozine çevirmek için gerekli olan reaksiyonu katalizleyen enzime fenilalanin hidroksilaz (PAH) enzimi adı verilir ve fenilketonüri hastalarında karaciğerde işlev gören bu enzim aktif değildir. Dolayısıyla vücutta birikmiş olan fenilalanin ve türevleri beyin omurilik sıvısına geçer, burada bileşiklerin düzeyi yükselir ve hasta bireyde zeka ve nörolojik gelişim geriliğine neden olur.
Rett sendromu, yaygın gelişimsel bozukluklardan birisi olarak sınıflandırılan beyinsel gelişim bozukluğudur. Ancak bunun yanlış bir sınıflandırma olduğunu ve benzer şekilde otistik belirtiler gösteren frajil X sendromu, tüberoz skleroz ya da Down sendromunun yaygın gelişimsel bozukluklar olarak sınıflandırılabileceğini önesüren görüşler bulunmaktadır. Bu sendromun belirtileri kolaylıkla otizm ve Angelman sendromunun belirtileriyle karışır. Klinik belirtiler arasında baş büyüme hızının azalması ve bazen mikrosefali, küçük el ve ayaklar bulunur. Stereotipik ve yineleyici el hareketleri de gözlenir. Bilişsel bozukluk ve gerileme döneminde de sosyalleşme sorunları da belirtiler arasında görülür. Okula girdikleri dönemde sosyalleşme genellikle düzelir. Rett sendromu olan kız çocuklar gastrointestinal bozukluklara yakalanmaya yatkındır ve %80’i nöbet geçirir. Hemen hemen hiç sözel becerileri yoktur ve kadınların %50’si yürüyemez. Skolyoz, büyüme eksikliği ve kabızlık çok yaygındır ve sorunlu olabilir.
Kistik fibrozis, akciğer, pankreas, bağırsak, ter bezleri dış salgı bezlerinde görülen, otozomal resesif kalıtımlı bir gen hastalığıdır. Kistik fibrozis hastalığı, aynı anda solunum sistemi, sindirim sistemi gibi vücudun birden çok sistem ve organını etkileyebilir. Doğumla birlikte görülen fibrozis, bu etkileme sonucu işlev bozukluklarına neden olur.

Erdheim Chester hastalığı ya da ECD, nadir rastlanan bir hastalık. Akyuvarın histiosit denilen özel bir türünün ya da makrofaj dokunun anormal çoğalması ile ayırt edilir. İlk kez, 1930 senesinde Amerikan patolog William Chester tarafından fark edilmiştir.(Teknik olarak, bu hastaklık Langerhans hücreli olmayan histiositozis olarak adlandırılır.) Hastalık, 2016 yılında Dünya Sağlık Örgütü tarafından histiositik neoplazm olarak ilan edildi. Başlangıcı tipik vakalarda olarak orta yaşta görülür Hastalık, lipid yüklü makrofajların, çok çekirdekli dev hücrelerin, kemik iliğinde inflamatuar bir lenfosit ve histiyosit sızıntısına ve uzun kemiklerin genelleştirilmiş bir sklerozuna sebep olur.
Hunter sendromu ya da Tip II mukopolisakkaridoz, iduronat-2-sülfataz (I2S) enziminin eksikliğinden ya da yokluğundan kaynaklanan lizozomal depo hastalığıdır. Doktor Charles A. Hunter (1873-1955) tarafından ilk kez 1917 yılında tanımlandığından, Hunter sendromu olarak adlandırılır.

Fabry hastalığı, nadir görülen X'e bağlı resesif kalıtılan lizozomal depo hastalığıdır. Geniş bir yelpazede sistemik semptomlara neden olabilir. Hastalık, ismini kaşiflerinden biri olan Johannes Fabry'den almaktadır.
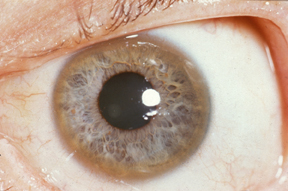
Wilson hastalığı veya hepatolentiküler dejenerasyon dokularda bakır birikimine yol açan otozomal resesif geçişli genetik bir hastalıktır. Bu hastalık kendini psikiyatrik veya nörolojik belirtilerle ve karaciğer hastalığıyla gösterir. Hastalığı ilaçla tedavi etmek mümkündür.

Orak hücreli anemi, alyuvarlardaki oksijen taşıyıcı protein olan hemoglobinin anormalliği sonucu alyuvarların orak şeklini almasıyla oluşan otozomal resesif kalıtılan genetik bir hastalıktır.
Reye sendromu, genellikle influenza (grip) veya suçiçeği gibi bir viral enfeksiyondan sonra özellikle aspirin alınmasıyla ortaya çıkar. Son yıllarda çocuklara ateş düşürücü olarak aspirin verilmemesi, doktorların bu konuda aileleri bilinçlendirmesi sonucu sıklığı giderek azalmıştır. Çoğu kez enfeksiyonun iyileşme döneminde aniden bulantı-kusma ve döküntülerle başlar, ancak aspirin alınmadan da ortaya çıkabilen vakalar vardır. Tam sebebi bilinmese de hücrelerin enerji santrali gibi çalışan mitokondrinin metabolik fonksiyonlarında bir bozulmaya bağlı olarak ortaya çıkar. Birçok organ etkilenmekle birlikte, karaciğer ve beyin en çok etkilenen organlardır. Karaciğerde genellikle yağlanma olur, ancak sarılık gibi karaciğer hastalığı belirtileri olmaz. Beyinde ciddi ödeme bağlı olarak şuur kaybıyla giden ağır bir ensefalopati tablosu görülür. Kalp, böbrek ve pankreasta da daha az seviyede yağlanma görülebilir. Erken tanı hayat kurtarıcıdır, şüphe olduğu anda, erkenden şuur kapanmadan önce yoğun bakım ünitesine alınmalı ve sıklıkla görülen kan şekeri düşüklüğüyle (hipoglisemi) ve beyin ödemiyle mücadele edilmelidir.
Çocukluk çağı alternan hemipleji, çocukluk çağı alternatifleşen- kısmi felci etiyolojisi bilinmeyen nadir bir nörolojik bozukluktur. Giderek çoğalan kanıtlar hastalığın öncelikli nedeni olarak ATP1A3 geninin değişinimini (mutasyonunu) göstermektedir. Atak olarak da ifade edilen, geçici ve bozukluktan muzdarip hemipleji nöbetleri ile bilinir. Hemipleji atakları vücudun bir ya da iki tarafında, hafif halsizlikten tamamen felce kadar, birçok şeye sebep olabilir. Süre olarak da değişken ve çeşitlidir. Ataklar vücudun bir tarafından diğerine, alternatif bir seyir izleyebilir. Tek atak esnasında, vücudun bir ya da iki tarafını etkilemek üzere değişebilir. Hastalık hemipleji ile birlikte birçok belirtiye sahiptir. Belirtilerin çoğu bebeklik döneminde ortaya çıkar. Bebek 18 aylıktan önce tipik belirtilere sahiptir. Genellikle hemipleji ve diğer ilgili belirtiler uyku ile kesilir, ancak uyanınca tekrarlanabilir. Bu nörolojik bozukluk yakın bir tarihte keşfedilmiştir. Karakteristik özellikleri 1971'de tanılanmıştır. 1 milyon insanda 1 birey olmak üzere çok nadir görülen bir hastalıktır. Hemipleji ile birlikte bozukluğun belirtileri çok geniş nörolojik ve gelişimsel zayıflıklar içerir. Bu hastalık kamuoyunda yeterince bilinmemektedir. Tüm dünyada 7.000 kişide bulunduğu tahmin edilirken, hastaların yalnızca %10'u STK ya da topluluklardan bilgi desteği almaktadır. Hastalık hakkındaki yayınların çoğu hastalık hakkındaki bilgi eksikliğini gidermeye yöneliktir.
Nörodejenerasyon, nöronların ölümü de dahil olmak üzere nöronların ilerleyen yapı veya fonksiyon kaybıdır. Nörodejeneratif süreçlerin bir sonucu olarak amiyotrofik lateral skleroz, Parkinson hastalığı, Alzheimer hastalığı, ölümcül ailesel uykusuzluk ve Huntington hastalığı gibi birçok nörodejeneratif hastalık ortaya çıkar. Bu tür hastalıklar tedavi edilemez ve nöron hücrelerinin ilerleyici dejenerasyonu ve / veya ölümüyle sonuçlanır. Araştırmalar ilerledikçe, bu hastalıkları hücre altı düzeyde birbirleriyle ilişkilendiren birçok benzerlik ortaya çıkmaktadır. Bu benzerliklerin keşfedilmesi, birçok hastalığı aynı anda iyileştirebilecek terapötik ilerlemeler için umut vermektedir. Atipik protein düzenekleri ve uyarılmış hücre ölümü dahil olmak üzere farklı nörodejeneratif bozukluklar arasında birçok paralellik vardır. Nörodejenerasyon, molekülerden sistemik olana kadar birçok farklı nöronal devre seviyesinde bulunabilir.

Polikistik böbrek hastalığı, böbreklerin görece sık karşılaşılan kistik hastalıklarındandır. 2 tip polikistik böbrek hastalığı vardır;
- Otosomal dominant polikistik hastalık
- Otosomal resesif polikistik hastalık