Hunter sendromu
| Hunter sendromu | |
|---|---|
| Uzmanlık | Endokrinoloji |
Hunter sendromu ya da Tip II mukopolisakkaridoz (MPS II), iduronat-2-sülfataz (I2S) enziminin eksikliğinden ya da yokluğundan kaynaklanan lizozomal depo hastalığıdır.[1][2] Doktor Charles A. Hunter (1873-1955) tarafından ilk kez 1917 yılında tanımlandığından, Hunter sendromu olarak adlandırılır.[3][4]
Genel bakış
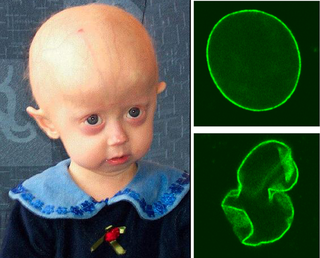
Hunter sendromu ya da mukopolisakkaridoz II (MPS II), öncelikle erkekleri etkileyen (X'e bağlı resesif) ciddi bir genetik bozukluktur. Bu hastalık vücudun dayanıklılık ve yenilenme kabiliyeti ile ilgili glikozaminoglikan (GAG) olarak da bilinen spesifik mukopolisakkaridozlarla ilgilidir. Hunter sendromu, lizozomal depo hastalıklarının ciddi tiplerinden birisidir. Hunter sendromunda, GAG'lar iduronat-2-sülfataz (I2S) enzim eksikliği ya da yokluğu nedeniyle vücutta hücrelerde birikirler. Bu birikim belli hücre ve organların vücut fonksiyonlarını etkiler ve bir dizi ciddi semptoma yol açar. GAG birikimi vücut hücrelerinde devam ettikçe, Hunter sendromunun belirtileri daha görünür hale gelir. Bu hastalığa sahip bazı insanlarda farklı yüz özellikleri ve büyük baş gibi fiziksel özellikler vardır. Hunter sendromu bazı durumlarda, merkezi sinir sistemi tutulumu, sinir sistemi sorunları ve gelişimsel gecikmelere yol açar. Hunter sendromu olan bütün insanlar tam olarak aynı şekilde etkilenmezler, semptomların progresyon oranı büyük ölçüde değişmektedir. Ancak, Hunter sendromu her zaman şiddetli, ilerleyici ve hayat sınırlayıcıdır.
Tanı
Genel olarak tanı yaşı 2-4 yaş arasındadır. MPS bozukluğunu, iduronat-2-sülfataz (I2S) enzim aktivitesi ölçümü ile kesin tanı koymadan önce yardımcı ek testler yapılır. MPS bozukluğu için en yaygın kullanılan laboratuvar tarama testi GAG için idrar testidir. İdrar testi GAG için bazen normal olabilir ama bu hastalık olmadığını göstermez. Hunter sendromunun kesin tanısı serumda, beyaz kan hücrelerinde ya da cilt biyopsisi ile fibroblastlar içinde I2S enzim aktivitesini ölçerek konulur. Hunter sendromlu bazı insanlarda I2S gen analizi ile klinik şiddet belirlenebilir. Prenatal tanı amniyotik sıvı veya koryonik villus dokusundan I2S enzimatik aktivitesini ölçerek konulabilir.
Semptom ve bulgular
En belirgin bulgu zeka geriliğidir; hafif formları ve şiddetli formları vardır. Diğer semptomlar
- Yüz değişiklikleri: kalın kaslar, düz, basık burun kökü, etli ve geniş dudaklar, büyük dil, prognathism.
- Derin ve boğuk ses
- Kardiyak tutulum ve kalp yetmezliği
- Orta kulak ve iç kulakta sağırlık
- Optik atrofi
- Erken başlangıçlı, ilerleyici eklem kontraktürleri,
- Hepatosplenomegali
- Göbek fıtığı
- Büyüme geriliği
- İskelet anomalileri (Dysostosis multipleks konjenita)
Ağır hastalarda, saldırganlık, kararsız yürüme ve sık düşme ile kaba motor bozukluklar ve son olarak bir süre sonra tetraspastik bozuklukları gelişir.
Genetik
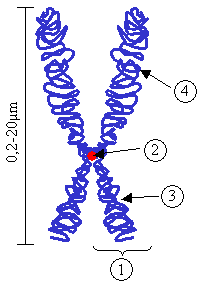
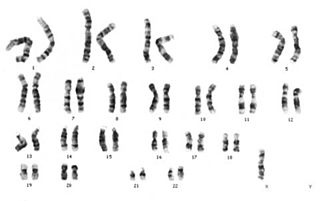
Hunter sendromu kalıtsal bir bozukluk olduğundan (X'e bağlı resesif, öncelikle erkekleri etkiler) belirli bir şekilde nesilden nesile aktarılır. İnsan vücudundaki neredeyse her hücre her bir ebeveynden gelen 23 kromozomdan oluşan toplam 46 kromozoma sahiptir. I2S geni X kromozomu üzerinde yer almaktadır. Erkeklerde bir X kromozomu varken dişilerde iki X kromozomu vardır. Erkekler X kromozomunu annesinden Y kromozomunu babasından alır. Dişiler X kromozomlarından birisini annesinden diğerini babasından alır. I2S geninin anormal bir kopyasına sahip bir erkek varsa onda Hunter sendromu gelişebilir. Bir erkek, iki yoldan biriyle I2S geninin anormal bir kopyasını alabilir. Birincisi annesi taşıyıcı olan (yani; bir anormal ve bir normal I2S geni taşıyan) kişiye anormal gen geçebilir. Alternatif olarak, yumurta ve sperm oluşumu sırasında X kromozomu üzerinde I2S geninde bir mutasyon gelişebilir. Bu ikinci durumda anne taşıyıcı değildir. Dişiler I2S geninin anormal bir kopyasını sadece taşırlar ve genellikle etkilenmezler.
Kaynakça
- ^ Wraith JE, Scarpa M, Beck M; ve diğerleri. (Mart 2008). "Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy". Eur. J. Pediatr. 167 (3). ss. 267-77. doi:10.1007/s00431-007-0635-4. PMC 2234442 $2. PMID 18038146.
- ^ James, William D.; Berger, Timothy G.; ve diğerleri. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ^ "Hunter's syndrome (Charles A. Hunter)". whonamedit.com. 10 Eylül 2012 tarihinde kaynağından arşivlendi. Erişim tarihi: 3 Şubat 2012. (İngilizce)
- ^ C. A. Hunter. A rare disease in two brothers. Proceedings of the Royal Society of Medicine, London, 1917, 10: 104-116.
Dış bağlantılar
- MPSsociety.org10 Ocak 2012 tarihinde Wayback Machine sitesinde arşivlendi., National MPS Society
- About.com29 Ocak 2012 tarihinde Wayback Machine sitesinde arşivlendi., "Hunter Syndrome", About.com
- Hunterpatients.com31 Mayıs 2020 tarihinde Wayback Machine sitesinde arşivlendi.
- NLM.NIH.gov2 Haziran 2010 tarihinde Wayback Machine sitesinde arşivlendi., GeneReview/NIH/UW entry on Mucopolysaccharidosis Type II
- HideandSeek.org29 Mayıs 2020 tarihinde Wayback Machine sitesinde arşivlendi., Foundation For Lysosomal Disease Research
- Elaprase.com4 Şubat 2012 tarihinde Wayback Machine sitesinde arşivlendi., (idursulfase) website.
- ClinicalTrials.gov12 Ağustos 2009 tarihinde Wayback Machine sitesinde arşivlendi.
- LysosomalStorageResearch.ca, The Hunter Disease eClinic and eBook - virtual education (İngilizce), (Japonca)
| Sınıflandırma | |
|---|---|
| Dış kaynaklar |