Horner sendromu
| Horner sendromu | |
|---|---|
 | |
| Sol taraflı horner | |
| Uzmanlık | Nöroloji |
Horner sendromu, Claude Bernard-Horner sendromu, Claude-Bernard-Horner sendromu veya Bernard-Horner sendromu, sempatik sinir sistemi hasarının neden olduğu klinik bir sendromdur.[1]
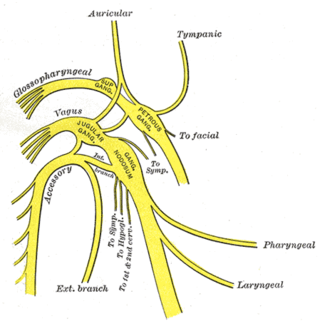
Pupil, üst göz kapağı, fasial ter bezleri ve fasial damarlara giden sempatik liflerde lezyon biçiminde açıklanan Horner sendromu, anhidrozis, miyozis, enoftalmus ve pitozis semptomları ile belirginleşir. Çocuklarda heterokromi (iki göz renginin farklı olması) görülebilir. Sempatik inervasyonda sorun olan göz daha açık renkli olur. Miyosizin sebebi m. dilatatör pupillanın denervasyonudur. Pitozisin sebebi m. tarsalis süperior ve m. levator palpabralis superiorun denervasyonudur. Ciliospinal refleks kaybolur. Bu refleks ipsilateral boyun veya yüz bölgesine verilen ağrılı uyarı ile ipsilateral midriyasiz olmasıdır. Yüzde flashing görülebilir. Sempatik denervasyon nedeni ile vazodilatasyon gelişir.[2]
Horner sendromunun nedenlerinden biri apeks tümörüdür. Genellikle akciğer kanserlerinin stellat gangliona yaptığı metastazın simpatik sinir liflerinde yaptığı hasara bağlı gelişmesine karşın; akciğer kanseri dışında bazı beyin içi lezyonlarda, kafa sinirleri ile ilgili patolojilerde, omurilik zedelenmelerinde, komada ve boyun sempatik sinir ağı yakınındaki diğer problemlerde ve az da olsa ciddi boğaz enfeksyonlarında görülebilir.[3] Horner sendromunun nadir sebeplerinden biri de tüp torakostomi sırasında drenin torakal sempatik gangliyonlara temas etmesidir. Reversible ya da irriversible olabilir. Tanısında kokain testinin önemi büyüktür.
Nedenler
- Periferik nedenler: travma (kaza, cerrahi), mediasten tümörleri (bronş ca, Hodgkin lenfoması, metastatik tümörler), tiroid adenomu, parafaringeal abse ya da tümör invazyonu, nörofibromatozis, boyun atardamarı (a.carotis interna) anevrizması, Pancoast sendromu[4][5][6]
- Santral siniri sistemi kökenli nedenler: beyin atardamarı (a.cerebralis) tıkanması, multipl skleroz (MS), siringomiyeli, beyin sapı tümörleri, beyin sapı lezyonları, omurilik (m.spinalis) servikal bölüm tümörleri, m.spinalis servikal bölüm travması, okülosempatik defekt, brakiyal pleksus travması, beyinde karotis kökenli iskemi, migren, orta beyin tümörü, çocuklarda sol sürrenaldeki nöroblastoma metastazı[4][5][6][7]
Bulgular
- Endoftalmi (nöroblastomanın orbita metastazlarında unilateral ekzoftalmi)
- Ptozis
- Myozis
- Disartri
- Disfaji
- Baş dönmesi
- Bulantı
- Etkilenen yüz yarısında terleme azalması
- N. trigeminus trajesinde parestezi
Kaynakça
- ^ Costopoulos, C.; Patel, R. S.; Mistry, C. D. (Mayıs 2008). "Painful Horner's syndrome". Emergency medicine journal: EMJ. 25 (5): 295. doi:10.1136/emj.2007.051953. ISSN 1472-0213. PMID 18434470. 26 Şubat 2024 tarihinde kaynağından arşivlendi. Erişim tarihi: 26 Şubat 2024.
- ^ Fetzer, S. J. (Nisan 2000). "Recognizing Horner's syndrome". Journal of Perianesthesia Nursing: Official Journal of the American Society of PeriAnesthesia Nurses. 15 (2): 124-128. doi:10.1053/pa.2000.5787. ISSN 1089-9472. PMID 11111529. 26 Şubat 2024 tarihinde kaynağından arşivlendi. Erişim tarihi: 26 Şubat 2024.
- ^ Sharma, Vaishnavi S.; Yadav, Vaishnavi (Aralık 2023). "Effect of Prehabilitation in Lung Cancer Patients Undergoing Lobectomy: A Review". Cureus. 15 (12): e49940. doi:10.7759/cureus.49940. ISSN 2168-8184. PMID 38179388.
- ^ a b Amonoo-Kuofi HS. Horner’s Syndrome revisited: with an update of the central pathway, Clinical Anatomy, 12:345–361, 1999
- ^ a b Reede DL, Garcon E, Smoker WR, Kardon R. Horner's syndrome: clinical and radiographic evaluation. Neuroimaging Clinics of North America, 18(2):369-385, 2008
- ^ a b Gonzalez Martín-Moro J, Sastre-Perez J, Pena Fernandez I. Horner syndrome after temporomandibular joint arthroscopy: a new complication. Journal of Oral & Maxillofacial Surgery, 67(6):1320-1322, 2009
- ^ Barrea C, Vigouroux T, Karam J, et al. Horner syndrome in children: A clinical condition with serious underlying disease. Neuropediatrics, 47(4):268-272, 2016
| Sınıflandırma | |
|---|---|
| Dış kaynaklar |