Guillain-Barré sendromu (GBS), çevresel sinir sisteminin edinilmiş bir bağışıklık kökenli yangısal bozukluğudur; merkezi sinir sistemi etkilenmez. Bu hastalık için kullanılan diğer isimler şöyledir: akut enflamatuvar demiyelinize edici polinöropati, akut idiyopatik poliradikülonörit, akut idiyopatik polinörit, Fransız polyosu, Landry'nin yükselici felci.

Huzursuz bacak sendromu (HBS), uyku ya da istirahat esnasında bacaklarda hissedilen rahatsızlık, huzursuzluk, hareket ettirme ihtiyacı, uyuşma, karıncalanma bazen de tam olarak tanımlanamayan bir histir.

Down sendromu, trizomi 21 ya da mongolizm; genetik düzensizlik sonucu insanın 21. kromozom çiftinde fazladan bir kromozom bulunması durumu ve bunun sonucu olarak ortaya çıkan genetik bir bozukluktur. Down sendromu, bireyin 1 yaşından daha uzun süre yaşayabildiği tek otozomal trizomidir.
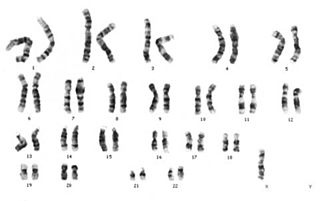
Turner sendromu, kromozom anomalisi sonucu ortaya çıkan ve kız çocuklarını etkileyen bir sendromdur; bir dişide eşey kromozomlarından birinin bulunmaması sonucu ortaya çıkar. Turner sendromluların fenotipi dişi olarak görülür fakat; eşey organları ve eşey hücreleri gelişmez. Kısır bireylerdir.
Sendrom ya da belirgi, birbirleriyle ilişkisiz gibi görünen, ancak bir araya geldiklerinde tek bir olgu olarak kendilerini gösteren bulgular bütünüdür. Sendromlar, kalıtsal olabilir ya da edinsel nedenlerle de oluşabilir. Kalıtsal sendromlar otosomal dominant, otosomal resesif ya da X-kromozomu aracılığıyla aktarılırlar; bir bölümünün nedeni bilinmemektedir. Sendromlar bir hastalık tablosunun gelişmesi içinde bir bulgu olabildiği gibi, bazı hastalıklar bazı sendromların komplikasyonu olarak belirlenirler.
Polikistik over sendromu (PCO), yumurtalıklarda birçok küçük iyi huylu kist oluşmasıyla beliren bir hastalıktır. Yumurtalıkta oluşan ve kist olarak adlandırılan bu organizmalar yumurtalıkların çevresine yerleşmiş çok sayıda yumurta hücresidir. Bu hücreler ultrasonda özel bir görüntü oluşturmaktadırlar.
Rett sendromu, yaygın gelişimsel bozukluklardan birisi olarak sınıflandırılan beyinsel gelişim bozukluğudur. Ancak bunun yanlış bir sınıflandırma olduğunu ve benzer şekilde otistik belirtiler gösteren frajil X sendromu, tüberoz skleroz ya da Down sendromunun yaygın gelişimsel bozukluklar olarak sınıflandırılabileceğini önesüren görüşler bulunmaktadır. Bu sendromun belirtileri kolaylıkla otizm ve Angelman sendromunun belirtileriyle karışır. Klinik belirtiler arasında baş büyüme hızının azalması ve bazen mikrosefali, küçük el ve ayaklar bulunur. Stereotipik ve yineleyici el hareketleri de gözlenir. Bilişsel bozukluk ve gerileme döneminde de sosyalleşme sorunları da belirtiler arasında görülür. Okula girdikleri dönemde sosyalleşme genellikle düzelir. Rett sendromu olan kız çocuklar gastrointestinal bozukluklara yakalanmaya yatkındır ve %80’i nöbet geçirir. Hemen hemen hiç sözel becerileri yoktur ve kadınların %50’si yürüyemez. Skolyoz, büyüme eksikliği ve kabızlık çok yaygındır ve sorunlu olabilir.
Cushing sendromu, 1932 yılında Harvey Williams Cushing tarafından tanısı ilk kez konmuş olan kortizol hormonunun olağanın üstünde bir düzeyde olduğu durumlarda ortaya çıkan belirtiler bütünüdür. Diğer isimleri Itsenko-Cushing sendromu, hiperadrenokortisizm veya hiperkortisizm. Cushing sendromunun alışılmış nitelikleri kilo artması, obezite, kan basıncının artması (hipertansiyon) ve derinin zayıflaması sonucu oluşan çizgilerdir. Her hastada belirtilerin tümü gözlenmeyebilir. Belirtilerin ağırlığı ve niceliği hastanın ne denli uzun bir süredir kortizol etkisinde kalmasına bağlıdır. Ancak kimi belirtiler bu durumdan bağımsız, iveğen olarak da gelişebilir. Örneğin, özellikle uyluk kemiğinin baş bölgesinin iveğen doku ölümüne uğraması çok kısa bir süredir hafif izleyen ya da yıllardır ağır izleyen Cushing sendromlu hastalarda da rastlanabilir.

Superior vena kava sendromu (SVCS) superior vena kavanın kompresyonu sonucunda ortaya çıkan belirti ve bulguları tanımlar. Superior vena kava sendromuna, trakea basısı da eşlik ederse superior mediastinal sendrom adını alır.
Hipoplazi ya da az gelişim, bir organın yetersiz gelişme nedeniyle doğumsal olarak küçük kalmasıdır. Organ, tüm anatomik özelliklerini taşır, fizyolojik işlevlerini yapabilir. Körelme, irileşim ve aşırı gelişim az gelişimin (hipoplazi) karşıtı olan kavramlardır; bu olguların tümü edinseldir. Hipoplazi kavramına en yakın olan olgu aplazi olgusudur; hipoplazi ve aplazi doğumsal (konjenital) patolojilerdir.
Hasta hakları, hastaların sağlık kurumları ve sağlık çalışanlarıyla ilişkilerinde bir insan ve hasta olarak sahip olduğu haklar bütünüdür.
Diş hekimliğinde, hipodonti edinsel ya da doğumsal diş eksiklikleri olgusu için kullanılan terimlerdendir; anodonti ve oligodonti kavramları da hipodonti başlığı altında yer alan diş eksikliği olgularıdır.

Radyasyon hastalığı olarak da bilinen Akut Radyasyon Sendromu (ARS), kısa bir süre boyunca yüksek miktarlarda iyonlaştırıcı radyasyona maruz kalınması nedeniyle ortaya çıkan sağlık etkileridir. İlk günlerde semptomlar mide bulantısı, kusma ve iştahsızlığı olabilir. Bunu birkaç saatliğine ya da haftalığına küçük semptomlar takip edebilir. Bundan sonra, toplam radyasyon dozuna bağlı olarak, insanlarda enfeksiyon, kanama, dehidrasyon ve şaşkınlık gelişebilir veya az semptomlu olarak geçirilebilir. Son olaraksa bunu ölüm ya da iyileşme izler. Belirtiler bir saat içinde başlayabilir ve birkaç ay sürebilir.
Hipertelorizm organizmada simetrik olarak bulunana çift organlar arasındaki uzaklığın normalden fazla olmasıdır. Ancak, günümüzde “hipertelorizm” tek başına kullanıldığında “gözler arasındaki mesafenin normalden fazla olması” olarak anlaşılır.
Ektodermal displazi sendromları, ektodermden gelişen doku ve organlardaki malformasyonların ve oluşum kusurlarının saptandığı geniş bir topluluktur. Dişlerle ilgili anomaliler ve malformasyonlar; saç oluşumunda yetersizlikler (seyrek/kırılgan/oluşmama); ter bezlerinin eksikliğine bağlı terleme azlığı/yokluğu (hipohidroz); başkaca çeşitli deri ve tırnak patolojileri ortak bulgular olarak saptanır. Günümüze dek ektodermal displazi bulgularını içeren 200'e yakın sendrom bildirilmiştir; bu kadar çok sayıda fenotip bulunmasının nedeni farklı genlerde oluşan mutasyonlardır. Ektodermal displazili ailelerdeki kalıtım otosomal dominant, otosomal resesif ya da x-kromozomu aracılığıyla resesif olarak aktarılır.
Mikrognati (micrognathism), altçenenin yetersiz gelişmesi (hipoplazi) niteliğinde bir anomalidir; 1. ve 2. faringeal arklara (brankial yarık) özgü malformasyonların çoğu çene-yüz bölgesindeki mezenkimal dokunun embriyolojik dönemdeki yetersizliğine bağlanmaktadır. Altçene küçüktür ve geridedir; bu olgu, çevredeki kasların hareketlerini sınırlarken bu kaslardan yararlanan dokuların gelişmeleri de kısıtlanır. Kraniyofasiyal anomalileri içeren sendromlarda çok sık görülen bulgulardan biridir. Sendroma-özgü olmayan olguların varlığı da bilinmektedir; örneğin; altçenesi, kafa ile göğüs kafesi arasında sıkışmış olan bir fetüste çene kemiklerinin gelişimi olması gereken düzeye ulaşamamaktadır. Gebelik sorunları, gebelerin alkol kullanması, gestasyonel diabet (gebelik diabeti) gibi çevresel faktörler etkili olabilmektedir (Möbius sendromu). Olguların bir bömümü, gebelerdeki rubella (kızamıkçık) infeksiyonu komplikasyonudur. Yenidoğanların bir bölümünde görülebilen hafif mikrognatiler, altçene gelişiminin tamalanmasıyla silinebilir. Mikrognati'de altçene gövdesi kadar çene eklemini oluşturan yapıların hipoplazisi de önemlidir. Mikrognatilerin bir bölümü kulak sistemi, üstçene ve damak gelişiminin de aksadığı olgularla birlikte görülür.

Makrosefali , nitelemesi kafatasının aşırı büyüklüğünü tanımlar. Primer makrosefali olgularının büyük bölümü sendroma-özgü bir bulgu olarak ortaya çıkar; bazıları ise başka bir bulgu olmaksızın görülen “ailesel makrosefali”lerdir. Sekonder makrosefali olguları, bir hastalığın temel bulgusu ya da komplikasyonu olarak ortaya çıkarlar ; başka bir bölümü ise, kafatası suturaları kapanmadan önceki dönemlerde geçirilen infeksiyonlar, subdural ya da intraventriküler kanamalar gibi çevresel nedenlere bağlı olabilir. Makrosefali, hidrosefalisi olan çocuklardaki en tipik bulgulardan biridir. Primer ya da sekonder olgularda makrosefalinin neden olduğu nörolojik bulgular saptanabilir.

Kraniyofasiyal yarıklar, kraniyofasiyal malformasyonların en önemlilerinden biridir; baş-boyun ve yüz bölgesinin oluşma ve gelişme aşamalarındaki aksamalar ya da sapmalar sonucu ortaya çıkan yapısal ve işlevsel bozuklukların önemli bir bölümünü oluştururlar. Embriyolojik kökenlerine göre; nöral tüp kökenli anomaliler, 1. ve 2. farengeal ark kökenli malformasyonlar, ektodermal displaziler söz konusudur.
Aşırı büyüme sendromları , çocuklarda, doğum öncesi (prenatal) dönemde ya da doğumdan sonra (postnatal) dönemde ortaya çıkabilen, bebeklerin/çocukların vücutlarının tümünde ya da bir bölümünde ortaya çıkan irileşmeyle karakterize olgulardır; aşırı büyümelerin görüldüğü sendromlarda, kilo ve boy artışı, çeşitli anomaliler ile zeka geriliği ve tümör riskinin varlığı en sık rastlanan bulgulardır.
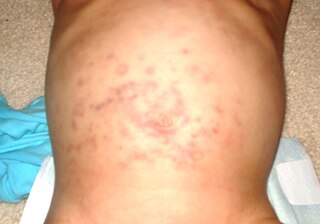
Stevens-Johnson sendromu, cildi, mukoza zarını, cinsel organları ve gözleri etkileyen nadir fakat ciddi bir hastalıktır.