
Genetik ya da kalıtım bilimi, biyolojinin organizmalardaki kalıtım ve genetik varyasyonu inceleyen bir dalıdır. Türkçeye Almancadan geçen genetik sözcüğü 1831 yılında Yunanca γενετικός - genetikos ("genitif") sözcüğünden türetildi. Bu sözcüğün kökeni ise γένεσις - genesis ("köken") sözcüğüne dayanmaktadır.
Mutasyon ya da değişinim, bir canlının genomu içindeki DNA ya da RNA diziliminde meydana gelen kalıcı değişmelerdir. Mutasyona sahip bir organizma ise mutant olarak adlandırılır.

Kromozom 8, 22 çift otozomal insan kromozomlarından 8. olanıdır. İnsanlarda normalde bir çift halinde bulunur. 146 milyon baz çiftine ve toplam hücre DNA'sının %4,5 ya da %5'ine sahiptir. Kromozom 8, muhtemelen 700 ile 1,000 arasında gen içermektedir.
Kromozom 18, toplamda 22 çift olan otozomal insan kromozomlarından onsekizincisidir. İnsanlarda normalde bir çift halinde bulunur. 76 milyon baz çiftine ve toplam hücre DNA'sının %2,5'ine sahiptir. Kromozom 18, muhtemelen 300 ile 400 arasında gen içermektedir.
Kromozom 22, insan genomunda toplamda 23, otozom olaraksa 22 adet bulunan kromozom çiftlerinden biridir. Normal insan genomundaki tüm otozomlar için geçerli olduğu gibi, kromozom 22 de iki kopya halinde bulunur. İkinci en küçük insan kromozomu olan kromozom 22, yaklaşık 49 milyon baz çiftiyle, toplam hücre DNA'sının %1,5 - 2'sini kapsar.
Kromozom anomalileri; bir kromozomda meydana gelen yapısal ya da sayısal değişiklikleri gösterir. Genellikle mayoz ve mitozu izleyen hücre bölünmesi sırasında meydana gelen hatalardan kaynaklanırlar. Birkaç farklı türü bulunmakla beraber, genel olarak sayısal ve yapısal anomaliler olarak iki ana gruba ayrılırlar.

Down sendromu, trizomi 21 ya da mongolizm; genetik düzensizlik sonucu insanın 21. kromozom çiftinde fazladan bir kromozom bulunması durumu ve bunun sonucu olarak ortaya çıkan genetik bir bozukluktur. Down sendromu, bireyin 1 yaşından daha uzun süre yaşayabildiği tek otozomal trizomidir.
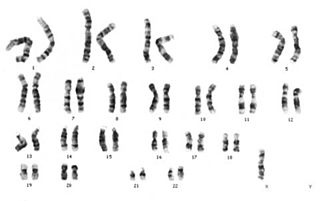
Turner sendromu, kromozom anomalisi sonucu ortaya çıkan ve kız çocuklarını etkileyen bir sendromdur; bir dişide eşey kromozomlarından birinin bulunmaması sonucu ortaya çıkar. Turner sendromluların fenotipi dişi olarak görülür fakat; eşey organları ve eşey hücreleri gelişmez. Kısır bireylerdir.

Delesyon, genetikte bir kromozomun bir parçasının kopup, kaybolmasıyla meydana gelen kromozom anomalilerindendir. Kopan parçadaki genler kaybolur yani eksilir ve ciddi genetik hastalıklara sebep olur.

Translokasyon, bir kromozomun kaybolan parçasının ya da kopan bir parçasının başka bir kromozoma yapışması şeklinde görülen kromozom anomalilerindendir.
Sendrom ya da belirgi, birbirleriyle ilişkisiz gibi görünen, ancak bir araya geldiklerinde tek bir olgu olarak kendilerini gösteren bulgular bütünüdür. Sendromlar, kalıtsal olabilir ya da edinsel nedenlerle de oluşabilir. Kalıtsal sendromlar otosomal dominant, otosomal resesif ya da X-kromozomu aracılığıyla aktarılırlar; bir bölümünün nedeni bilinmemektedir. Sendromlar bir hastalık tablosunun gelişmesi içinde bir bulgu olabildiği gibi, bazı hastalıklar bazı sendromların komplikasyonu olarak belirlenirler.

Crosover veya krossing over ya da parça değişimi mayoz bölünmenin profaz I evresinde görülen, çift halde bulunan kromozomların yaptığı parça değişimine verilen addır. Bunun sonucunda genetik rekombinasyon meydana gelir. Yani farklı kromozomlarda bulunan genlerin alelleri birbiriyle yer değiştirir.
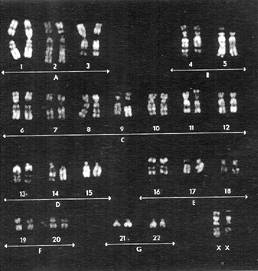
İzokromozom, herhangi bir kromozomun metafaz bölünmesi sırasındaki bir hata sonucu sentromerinden asimetrik biçimde bölünmesiyle oluşan anomalisine verilen addır.
Rett sendromu, yaygın gelişimsel bozukluklardan birisi olarak sınıflandırılan beyinsel gelişim bozukluğudur. Ancak bunun yanlış bir sınıflandırma olduğunu ve benzer şekilde otistik belirtiler gösteren frajil X sendromu, tüberoz skleroz ya da Down sendromunun yaygın gelişimsel bozukluklar olarak sınıflandırılabileceğini önesüren görüşler bulunmaktadır. Bu sendromun belirtileri kolaylıkla otizm ve Angelman sendromunun belirtileriyle karışır. Klinik belirtiler arasında baş büyüme hızının azalması ve bazen mikrosefali, küçük el ve ayaklar bulunur. Stereotipik ve yineleyici el hareketleri de gözlenir. Bilişsel bozukluk ve gerileme döneminde de sosyalleşme sorunları da belirtiler arasında görülür. Okula girdikleri dönemde sosyalleşme genellikle düzelir. Rett sendromu olan kız çocuklar gastrointestinal bozukluklara yakalanmaya yatkındır ve %80’i nöbet geçirir. Hemen hemen hiç sözel becerileri yoktur ve kadınların %50’si yürüyemez. Skolyoz, büyüme eksikliği ve kabızlık çok yaygındır ve sorunlu olabilir.

Gen duplikasyonu, içinde bir gen bulunan bir DNA bölgesinin herhangi şekilde ikilenmesidir; homolog rekombinasyon sırasında bir hata sonucu, retrotranspozisyon olayı veya tüm bir kromozomun ikilenmesi sonucu meydana gelebilir. Genin kopyası selektif baskıdan yoksun olduğu için, ondaki mutasyonların organizma üzerinde zararlı etkisi olmaz. Dolayısıyla, organizmanın nesilleri boyunca, işlevsel tek kopyalı bir gene kıyasla daha hızlı mutasyona uğrar.
Hunter sendromu ya da Tip II mukopolisakkaridoz, iduronat-2-sülfataz (I2S) enziminin eksikliğinden ya da yokluğundan kaynaklanan lizozomal depo hastalığıdır. Doktor Charles A. Hunter (1873-1955) tarafından ilk kez 1917 yılında tanımlandığından, Hunter sendromu olarak adlandırılır.
Konjenital bozukluk olarak da bilinen doğum kusuru, nedeni ne olursa olsun doğumda mevcut olan anormal bir durumdur. Doğum kusurları fiziksel, zihinsel veya gelişimsel engelliliklerle sonuçlanabilir.

Baskın veya dominantlık, genetikte bir genin karşılıklı lokuslar üzerinde bulunan alellerinden hangisinin canlının karakterini (fenotipini) belirleyeceğini gösteren ilişki.

Genetik hastalıklar , bir ailede kuşaktan kuşağa aktarılabilen patolojileri niteleyen tanımlamadır. Kalıtsal hastalıkların gelecek kuşaklara aktarılmasında etkili olan faktörlerler, genlerdeki ve kromozomlardaki yapısal değişikliklerdir.

Saf kırmızı hücre aplazisi (PRCA), kemiklerin merkezindeki süngerimsi doku olan kemik iliğinin yeterli şekilde işlev göremediği ve anemiye neden olduğu nadir görülen bir kan üretimi bozukluğudur. Kırmızı kan hücreleri, tüm vücuda oksijen taşımaktan sorumludur.