
Anemi, yani halk arasında bilinen adıyla "kansızlık", toplam kırmızı kan hücresi/alyuvar/Eritrosit sayısının azalması veya eritrositlerin içindeki hemoglobin miktarının azalması veya her ikisinin birlikte olması sonucu oluşan bir hastalıktır. Anemi ismi Grekçe: ἀναιμία Grekçe: anaimia, ἀν- an-, "-sız" + αἷμα haima, "kan" kelimelerinden türetilmiştir. Eritrositlerin içinde bulunan hemoglobinin, oksijeni akciğerlerden kapiller arterlere taşıması nedeniyle anemi hücre, doku ve organlarda hipoksiye neden olabilir. Oksijenin hücre canlılığı için elzem olması nedeniyle eksikliği pek çok klinik sonuca neden olur.

Trombosit veya kan pulcukları, kan pıhtılarının oluşumunda görev alan hücre parçalarına verilen isimdir. Platelet olarak da adlandırılır. Düşük trombosit seviyeleri veya fonksiyon anormallikleri (disfonksiyon) kanamaya yatkınlığı artırırken, yüksek trombosit seviyeleri -çoğunlukla asemptomatik- tromboz riskini yükseltir.

Karaciğer sirozu veya kısaca siroz, uzun süreli karaciğer hasarının neden olduğu, karaciğer fonksiyonunun yaygın ve çoğu zaman geri dönüşümsüz olarak bozulmasıyla karakterize kronik ve ilerleyici bir hastalıktır. Karaciğerde meydana gelen hasar sonucu zamanla normal karaciğer dokusunun yerini fibröz skar dokusu alır ve bu da karaciğer fonksiyonlarının bozulmasına yol açar. Hastalık tipik olarak aylar veya yıllar içinde yavaşça gelişir. Erken belirtiler arasında yorgunluk, halsizlik, iştah kaybı, açıklanamayan kilo kaybı, bulantı, kusma ve karnın sağ üst kadranında ağrı olabilir. Hastalık ilerledikçe belirtiler arasında kaşıntı, alt bacaklarda şişme, karında sıvı birikmesi, sarılık, kolay morarma ve ciltte örümcek benzeri kan damarlarının gelişmesi yer alabilir. Karında biriken sıvı spontan enfeksiyonlara yol açabilir. Daha ciddi komplikasyonlar arasında hepatik ensefalopati, özofagus, mide veya bağırsaklardaki genişlemiş damarlardan kanama ve karaciğer kanseri yer alır. Sirozun evreleri arasında kompanse siroz ve dekompanse siroz yer almaktadır.

Alzheimer hastalığı (AH), genellikle yavaş yavaş başlayan ve giderek kötüleşen nörodejeneratif bir hastalıktır ve demans vakalarının %60-70'inin nedenidir. En sık görülen erken belirti yakın zamanda yaşanan olayları hatırlamada zorluktur.
Trombotik trombositopenik purpura, kanın pıhtılaşmasına ilişkin nadir görülen bir hastalıktır. Vücut genelinde küçük damarlarda mikroskobik ve yoğun kan pıhtılaşmasıyla kendisini gösterir.
Hunter sendromu ya da Tip II mukopolisakkaridoz, iduronat-2-sülfataz (I2S) enziminin eksikliğinden ya da yokluğundan kaynaklanan lizozomal depo hastalığıdır. Doktor Charles A. Hunter (1873-1955) tarafından ilk kez 1917 yılında tanımlandığından, Hunter sendromu olarak adlandırılır.

Fabry hastalığı, nadir görülen X'e bağlı resesif kalıtılan lizozomal depo hastalığıdır. Geniş bir yelpazede sistemik semptomlara neden olabilir. Hastalık, ismini kaşiflerinden biri olan Johannes Fabry'den almaktadır.
Hipertrikoz, vücudun her yerindeki kılların anormal miktarda uzamasıdır. Hipertrikoz görünümü mitolojik kurt adama benzediğinden dolayı, yaygın olarak gayriresmi şekilde kurt adam sendromu olarak da adlandırılır. Vücudun pek çok yerinde oluşan jeneralize ve yalnızca belirli bir bölgede oluşan lokalize olmak üzere iki tür hipertrikoz bulunmaktadır. Hipertrikoz kişide doğuştan gelen veya sonradan gelişen bir hastalık olabilir. Androjene bağımlı kıl hariç olmak üzere kıllar cilt üzerinde, kasık bölgesinde, yüz ve koltuk altı bölgelerinde meydana gelir.
Kasabach - Merritt sendromu (KMS) ayrıca hemanjiom - trombositopeni sendromu olarak da bilinen genellikle bebekleri etkileyen nadir bir hastalıktır. Yaşamı tehdit edici bu vasküler tümör sendromunda azalmış trombosit sayısı ve bazen diğer kanama problemleri ile birlikte hemanjiom bulunur. Hastalık adını Haig Haigouni Kasabach ve Katharine Krom Merritt isminde, ilk kez bu durumu 1940 yılında tarif eden iki pediatristten almıştır.

Buruli ülseri Mycobacterium ulcerans'ın neden olduğu bulaşıcı hastalıktır. Hastalığın erken evresinde enfeksiyon, ağrısız bir nodül veya bölgesel şişlik ile karakterizedir. Bu nodül ülser'e dönüşebilir. Ülser içeride deri yüzeyindekinden daha büyük ve şişlik ile çevrili olabilir. Hastalık kötüleştikçe, kemiğe de bulaşabilir. Buruli ülserleri çoğunlukla kol veya bacakları etkiler; ateş yaygın değildir.

Ekvator frengisi ve pian olarak da bilinir. Spiroket bakterisi Treponema pallidum pertenue'nin yol açtığı tropik bir cilt, kemik ve eklem enfeksiyonudur. Hastalık ciltte 2 ila 5 santimetre yarıçapında yuvarlak ve sert kabarmalar ile başlar. Ortası patlayarak açılabilir ve ülsere neden olabilir. Bu ilk cilt lezyonu tipik olarak 3 ila 6 ayda iyileşir. Haftalar ve yıllar sonra, eklemler ve kemiklerde ağrılı yorgunluk ortaya çıkabilir ve yeni cilt lezyonları görülebilir. avuç içi ve ayakların tabanları kalınlaşabilir ve çatlayabilir. Kemiklerde şekil bozukluğu oluşabilir. Beş yıl ve daha sonra büyük ölü cilt bölgeleri ve ardından yara izleri görülebilir.
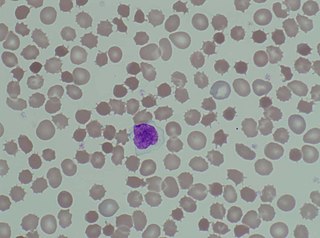
Biyoloji ve tıpta, akantosit hücre zarında anormal dikenli projeksiyonlar içeren kırmızı kan hücresi biçimini ifade eder. Ekinosit veya şistositlerle karıştırılabilirler.
Werner sendromu (WS), aynı zamanda "yetişkin progeriası" olarak da bilinir, erken yaşlanmanın ortaya çıkmasıyla karakterize, nadir görülen, otozomal resesif geçişli bir genetik hastalıktır.
Psoriatik artrit, sedef hastalığından etkilenen kişilerde ortaya çıkan uzun süreli bir inflamatuar artrittir. Psoriatik artritin klasik özelliği, sosis benzeri bir görünümle tüm el ve ayak parmaklarının şişmesidir. Bu genellikle, tırnaktaki küçük çöküntüler (çukurlaşma), tırnakların kalınlaşması ve tırnağın tırnak yatağından ayrılması gibi tırnaklardaki değişikliklerle birlikte olur. Sedef hastalığı ile uyumlu cilt değişiklikleri sıklıkla psöriatik artritin başlangıcından önce meydana gelir, ancak etkilenen bireylerin %15'inde psöriatik artrit döküntüden önce gelebilir. Bir tür seronegatif spondiloartropati olarak sınıflandırılır.
Otoimmün hastalık, bir vücut kısmına anormal bir bağışıklık tepkisinden kaynaklanan bir durumdur. En az 80 otoimmün hastalık türü tanımlanmış olup, bazı kanıtlar 100'den fazla türün olabileceğini düşündürmektedir. Herhangi bir vücut parçası tutulabilir. Semptomlar çeşitlidir ve genellikle hafif ila şiddetli arasında değişen ve geçici olabilen düşük dereceli ateş ve yorgun hissetmeyi içerir.
Marinesco–Sjögren sendromu (MSS), bazen Marinescu-Sjögren sendromu olarak da adlandırılır, nadir görülen otozomal resesif bir hastalıktır.
Hiperpigmentasyon, artan melanin nedeniyle cilt veya tırnak bölgesinin koyulaşmasıdır.
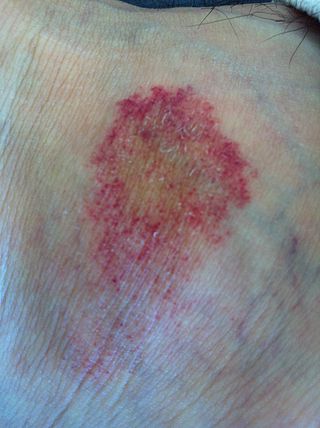
Pigmente purpurik dermatoz, purpurik deri erüpsiyonlarıyla (döküntü) karakterize deri durumlarının üç büyük sınıfından birisini ifade eder.
Eozinofilik folikülit (EF), bilinmeyen bir neden dolayı ortaya çıkan bir kaşınan kızarıklıktır. Bu hastalık en çok HIV enfeksiyonu olan kişilerde görülür ancak HIV negatif kişilerde de ortaya çıkabilir, bu durumda eponim bir hastalık olan Ofuji hastalığı olarak bilinir. Eozinofilik folikülitte saç foliküllerinde kaşınan kırmızı şişlikler (papül) görülür, eozinofilik folikülit karnı ve bacakları etkilememekle birlikte üst vücudu tutar. Eozinofilik folikülit adı, hastalıkta görülen en belirgin bağışıklık sistemi hücreleri (eozinofil) ve saç folikülü tutulumunu ifade eder.

Stratum granülozum epidermisin stratum spinozum katmanının üzerinde ve stratum korneum katmanının altında yer alan ince bir hücre katmanıdır. Alttaki stratum spinozumdan yukarı çıkan keratinositler, stratum granülozumda granüler (tanecikli) hücreler olarak bilinir. Granüler hücrelerde keratin filamentlerini birbirlerine bağladığı düşünülen histidin ve sistein miktarınca zengin proteinleri içeren keratohyalin granülleri bulunur; bu nedenle keratohyalin granüllerinin temel işlevi, ara keratin filamentlerini birbirlerine bağlamaktır.
