
Genetik ya da kalıtım bilimi, biyolojinin organizmalardaki kalıtım ve genetik varyasyonu inceleyen bir dalıdır. Türkçeye Almancadan geçen genetik sözcüğü 1831 yılında Yunanca γενετικός - genetikos ("genitif") sözcüğünden türetildi. Bu sözcüğün kökeni ise γένεσις - genesis ("köken") sözcüğüne dayanmaktadır.
Mutasyon ya da değişinim, bir canlının genomu içindeki DNA ya da RNA diziliminde meydana gelen kalıcı değişmelerdir. Mutasyona sahip bir organizma ise mutant olarak adlandırılır.

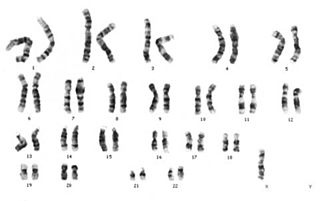
Kromozom 6, 22 çift otozomal insan kromozomlarından 6. olanıdır. İnsanlarda normalde bir çift halinde bulunur. Yaklaşık olarak 170 milyon baz çifti ve toplam hücre DNA'sının %5,5 ya da %6'sına sahiptir. Kromozom 6, muhtemelen 1,100 ile 1,600 arasında gen içermektedir.

X kromozomu, iki eşey kromozomundan biridir.
Y kromozomu, iki eşey kromozomundan biridir. Bütün erkeklerde normalde "44, XY" normal karyotip olarak X kromozomuya birlikte bulunur.
Turner sendromu, kromozom anomalisi sonucu ortaya çıkan ve kız çocuklarını etkileyen bir sendromdur; bir dişide eşey kromozomlarından birinin bulunmaması sonucu ortaya çıkar. Turner sendromluların fenotipi dişi olarak görülür fakat; eşey organları ve eşey hücreleri gelişmez. Kısır bireylerdir.

Klinefelter sendromu ya da 47, XXY sendromu; hücre bölünmesi sırasında, eşeysel kromozom düzensizliklerinden kaynaklanan semptomların kişide görülmesi durumudur.
XYY sendromu veya süper erkek sendromu; erkek bireyin iki Y kromozomu taşımasına neden olan bir eşey kromozomlarında meydana gelen anöplodi durumudur.

Translokasyon, bir kromozomun kaybolan parçasının ya da kopan bir parçasının başka bir kromozoma yapışması şeklinde görülen kromozom anomalilerindendir.

Kedi miyavlaması sendromu, Kedi çığlığı sendromu veya tıptaki isimleriyle Cri du Chat sendromu ya da Cri-du-Chat sendromu, 5. kromozomun bir parçasının kaybıyla ilişkili nadir bulunan bir genetik düzensizliktir. Sendromun genetik tanımı 45,X(X/Y),-5p olarak gösterilir. Yani kişide 45 kromozomun bulunduğunu fakat 5. kromozomun kısa (petit) kolunun bir kısmının bulunmadığını ifade eder. Bu tip kromozom mutasyonlarında DNA'daki bazın ya da bazların yok olmasına delesyon adı verilir. Delesyondaki büyüklük bebeklerdeki fiziksel, psikomotor ve zihinsel gelişimlerini etkiler.
Huntington hastalığı (HH), genetik, nörodejeneratif bir beyin hastalığıdır. Hastalarda bazı hareket bozukluklarının yanı sıra mental gerilik görülür. Adını 1872 yılında hastalığın kalıtsal olduğunu ilk olarak gözlemleyen Dr. George Huntington'dan alır. Otozomal dominant olarak kalıtılan bir hastalık olup beyin ve sinir sistemini etkiler. Hasta kişiler genellikle heterozigotlardır. Hastalığın ilk belirtileri 30-50 yaş arasında gözlenir. Hasta bu dönem zarfında çocuk yapmış ise çocuklarına bu hastalığı aktarma riski %50 oranındadır. Hastalık ölümcül olmasa da bu durum hastalığın gelişimine bağlıdır. Hastalığın nedeni Huntington proteinin üretim bozukluğundan dolayı ortaya çıkmaktadır.
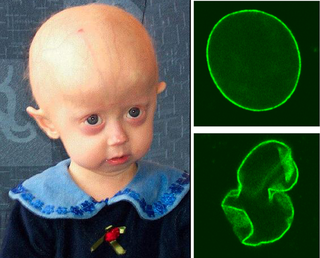
Progeria, halk dilindeki adıyla erken yaşlanma hastalığı, konu hakkında yapılan bilimsel araştırmalar hastalığın çaresini bulmaktan ziyade hastalığa sebep olan faktörleri bulmak ve bu sayede insanlığın ömrünü uzatabilmektir.
Genetik bozukluk, genlerde ve kromozomlarda görülen anomaliler sonucu ortaya çıkan durumdur. Kanser gibi bazı hastalıklar yaşam sırasında edinilen ve bazı hücrelerde görülen genetik anomaliler nedeniyle olsa da "genetik hastalık" terimi genellikle vücuttaki tüm hücrelerde bulunan ve döllenmeden beri var olan hastalıklar için kullanılır. Bazı genetik bozukluklar, sperm ve yumurtalar gibi üreme hücrelerini oluşturan mayoz bölünme sırasında oluşan kromozom anomalileri nedeniyle ortaya çıkar. Bunlara örnek olarak Down sendromu, Turner sendromu (45X0) ve Klinefelter sendromu sayılabilir. Diğer genetik değişiklikler ebeveynler tarafından tohum hücrelerin oluşturulması sırasında ortaya çıkabilir. Bunlara bir örnek frajil X sendromu ya da Huntington hastalığına neden olabilen üçlü yayılma tekrar mutasyonudur. Hatalı genler ebeveynlerden olduğu gibi alınmış da olabilir. Bu genellikle sağlıklı ama resesif gen taşıyan iki kişinin üremesi ya da hatalı genin dominant olması sonucunda olabilir.
Rett sendromu, yaygın gelişimsel bozukluklardan birisi olarak sınıflandırılan beyinsel gelişim bozukluğudur. Ancak bunun yanlış bir sınıflandırma olduğunu ve benzer şekilde otistik belirtiler gösteren frajil X sendromu, tüberoz skleroz ya da Down sendromunun yaygın gelişimsel bozukluklar olarak sınıflandırılabileceğini önesüren görüşler bulunmaktadır. Bu sendromun belirtileri kolaylıkla otizm ve Angelman sendromunun belirtileriyle karışır. Klinik belirtiler arasında baş büyüme hızının azalması ve bazen mikrosefali, küçük el ve ayaklar bulunur. Stereotipik ve yineleyici el hareketleri de gözlenir. Bilişsel bozukluk ve gerileme döneminde de sosyalleşme sorunları da belirtiler arasında görülür. Okula girdikleri dönemde sosyalleşme genellikle düzelir. Rett sendromu olan kız çocuklar gastrointestinal bozukluklara yakalanmaya yatkındır ve %80’i nöbet geçirir. Hemen hemen hiç sözel becerileri yoktur ve kadınların %50’si yürüyemez. Skolyoz, büyüme eksikliği ve kabızlık çok yaygındır ve sorunlu olabilir.
Hunter sendromu ya da Tip II mukopolisakkaridoz, iduronat-2-sülfataz (I2S) enziminin eksikliğinden ya da yokluğundan kaynaklanan lizozomal depo hastalığıdır. Doktor Charles A. Hunter (1873-1955) tarafından ilk kez 1917 yılında tanımlandığından, Hunter sendromu olarak adlandırılır.
Aicardi sendromu, beyindeki ana bağlantı yolu olan corpus callosum'un kısmi ya da tam yokluğu, retina anomalileri, infantil spazm şeklinde nöbetlerle karakterize nadir görülen genetik doğumsal hastalıktır. X kromozomuna bağlı olarak geçer, sadece kızlarda görülür ya da Klinefelter sendromu olan erkeklerde görülür.
Haplogrup, benzer haplotip gruplarının tümünde ortak atadan gelen aynı tek nükleotid polimorfizmi (SNP) mutasyonuna sahip gen serilerinin oluşturduğu grup. Bir haplogrup benzer haplotiplerden oluşur ve bu sayede bir haplogrubu haplotiplerinden tahmin etmek mümkündür. SNP testi bir haplogrubu doğrular. Haplogruplar harflerle adlandırılır. örneğin R1b1 çeşitli sayı ve harf kombinasyonlarından oluşur. Y kromozomu ve mitokondriyal DNA haplogrupları farklı haplogrupları temsil eder. Haplogrupların oluşumları binlerce yıl öncesine dayanmaktadır.

XXYY sendromu, erkeklerin fazladan bir X ve Y kromozomlarının bulunduğu eşey kromozomları bozukluğu. İnsan hücreleri genellikle anne ve babadan olmak üzere iki cinsiyet kromozomu içerir. Genellikle dişiler iki X kromozomuna (XX), erkekler bir X bir Y kromozomuna (XY) sahiptir. Düzgün çalışan bir SRY geni en az bir Y kromozomunun ortaya çıkmasıyla erkek olur. Bu nedenle XXYY kromozomuna sahip olan insanlar normalde erkek olur. XXYY sendromlu erkekler 46 yerine 48 kromozoma sahiptir. Bu yüzden XXYY sendromu bazen 48, XXYY sendromu olarak yazılır. Yaklaşık her 18.000-40.000 erkek doğumunda bir XXYY sendromlu bireyin doğduğu tahmin edilmektedir.

Genetik hastalıklar , bir ailede kuşaktan kuşağa aktarılabilen patolojileri niteleyen tanımlamadır. Kalıtsal hastalıkların gelecek kuşaklara aktarılmasında etkili olan faktörlerler, genlerdeki ve kromozomlardaki yapısal değişikliklerdir.

Triple X sendromu veya Süper dişi sendromu; X kromozomları ayrılmamış yumurta ile X kromozomu taşıyan normal bir spermin döllenmesi sonucu oluşur. Bu hastalığın tanısı hamilelik döneminde oldukça zordur. Bu bireylerin boy uzunluğu normal bir dişiden uzundur. Öğrenme güçlüğü, zekâ geriliği ve kısırlık görülür. Hastalık her 1000 dişi bebekten 1'inde görülür. 3. X Kromozomu dişinin karyotipindeki normal dişi gelişimini sağlayan genetik hassasiyeti bozar. Zira normalde dişilerdeki iki X kromozomundan 1'i inaktifken bu bireylerde iki X kromozomu inaktiftir. 2n= 44+XXX olarak da ifade edilebilir.
