
Osteoskleroz, kemik dokusundaki yoğunluk artışını betimler.
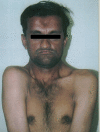
Cleidocranial dysostosis, kemik malformasyonları içeren kalıtsal bir sendromdur; ebeveynlerden birinden otosomal dominant yolla gelir. Genetik incelemeler, kemik gelişimiyle ilgili RUNX2 genindeki bir sorunu göstermektedir. 3 tipi vardır; dişlerin etkilenmesi bunlardan yalnızca birinde saptanır. İskelet sistemi etkilenmesinin en tipik bulgusu köprücük kemiklerinde (klavikula;clavicula) görülür. Köprücük kemikleri ya hiç oluşmamıştır (agenezis) ya da gelişmesi yetersizdir (hipoplazi); bu nedenle, hastalar, omuzlarını orta çizgi üzerinde birleştirebilirler. Kafatasındaki bıngıldaklar ve pelvisteki simfizi uzun süre kapanamaz.
Cardiofaciocutaneous sendrom (CFC), ektodermal displazi bulgularını da içeren, otosomal dominant geçen kalıtsal bir sendromdur. Klinik bulguları değişmeyen 4 fenotipi vardır. Bebeklerin çoğu prematüre doğar.

Coffin-Siris sendromu, ektodermal displazi bulgularını da içeren, otosomal dominant geçen kalıtsal bir sendromdur. 8 fenotipi vardır. Çene-yüz bulguları fenotiplerden 3'ünde belirgindir. Hastalarda genel gelişme geriliği vardır. Kafatası küçüktür (mikrosefali).
Kraniyoektodermal displazi (Cranioectodermal dysplasia), ektodermal displazi bulguları da içeren, otosomal resesif geçen kalıtsal bir sendromdur. 4 fenotipi vardır:
- Cranioectodermal dysplasia 1 (Sensenbrenner sendromu; Levin sendromu; arthrodentoosseous dysplasia)
- Cranioectodermal dysplasia 2
- Cranioectodermal dysplasia 3
- Cranioectodermal dysplasia 4
Oculodentodigital sendrom (ODDD; oculodentoosseous dysplasia), ektodermal displazi bulguları da içeren, otosomal dominant geçen kalıtsal bir sendromdur. Bazı olgular, otosomal resesif geçiş gösterebilir ya da ileri yaş gebeliklerden doğan bebeklerdeki spontan gen mutasyonunun sonucudur.
OPD sendromu (otopalatodigital sendrom), X-kromozomu aracılığıyla dominant (XLD) yolla geçen kalıtsal bir sendromdur (Gorlin-Cohen sendromu hariç). Kız çocukları daha hafif etkilenir. Tümü benzer bulgular içeren 5 tipi vardır:
- Otopalatodigital sendrom tip I (OPD tip1)
- Otopalatodigital sendrom tip II (OPD tip2)
- Gorlin-Cohen sendromu (ilgili sayfaya gidiniz)
- Melnick-Needles sendromu (tip 1 ile allelik bağlantısı vardır; ilgili sayfaya gidiniz)
- Terminal osseöz displazi ve pigmentli deri defektleri (çok enderdir, ayrıntı verilmemiştir).
Gorlin-Cohen sendromu , ektodermal displazi bulgularını da içerebilen bir OPD sendromu tipidir. 2 fenotipi vardır:
- Frontometaphyseal dysplasia 1
- Frontometaphyseal dysplasia 2
Melnick-Needles sendromu , X-kromozomu aracılığıyla dominant (XLD) yolla geçen kalıtsal bir sendromdur. Kız çocukları daha hafif etkilenir. Erkek çocuklarında güçlü OPD tip 2 bulguları saptanır; çoğu ölü doğar. Gelişme geriliği olabilir. Başlıca bulgular şunlardır:

Marshall sendromu, ektodermal displazi bulguları da içerebilen, otosomal dominant yolla geçen kalıtsal bir sendromdur.
Kraniyosinostoz, kraniyosinostozis (craniosynostosis), kraniyofasiyal malformasyonların ve maksillofasiyal sendromların önemli bir bölümünde etkileri görülebilen konjenital bir patolojidir. Bu olgudaki temel bulgu kafatası eklemlerinin erken kapanmasıdır; etkilediği anatomik bölgelerde ortaya çıkan malformasyonlar, hangi suturaların ne düzeyde kapanmış olmasıyla orantılıdır. Malformasyonlar genellikle etkilenen eklemin dikey yönünde belirgindir.

Cantu sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur.
Raine sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Yeni doğan ve bebek ölümleri sıktır; çoğu doğumdan sonraki haftalar içinde kaybedilen bebeklerde, kemiklerde yaygın yoğunlaşmalar (skleroz) saptanır. Özellikle kafa tabanı ve yüz kemiklerindeki skleroz oldukça belirgindir. Ayrıca, yoğun bir periostal kemik yapımı nedeniyle, uzun kemiklerin çevresinde sklerotik bir kılıf oluşur. Mikrosefali ve alın bombesi oldukça yüksek olan brakisefali (turribrakisefali) saptanır. Bıngıldaklar (fontaneller) tam kapanmamıştır. Kafatasındaki kemik yoğunlaşması (osteoskleroz) kraniyofasiyal displazi bulgularının oluşmasına yol açar. Ekzoftalminin yanı sıra hipertelorizm de olabilir. Kulaklar arkaya dönük ve biçimsizdir. Yüz orta bölümü hipoplazisi nedeniyle üstçene küçüktür (mikrognati), basık ve silik bir burun yapısı izlenir; bu yapı balık yüzü izlenimi verir. Altçene önde gibidir (prognatizm). Ağız açıklığı dardır. Derinliği fazla olan bir yarık damak varlığı beslenme bozukluklarına neden olur. Doğumda dişler olabilir, zamanında süren dişlerse küçüktür (mikrodonti). Hastaların büyük bölümünde dişeti büyümeleri vardır. Dil oldukça büyüktür (makroglossi).

Robinow sendromu, 5 fenotipi olan kalıtsal bir sendromdur; fenotiplerin ikisi otosomal resessif, üçü otosomal dominant yolla aktarılır. Tüm fenotiplerde iskelet sistemi bulguları yoğun olmakla birlikte maksillofasiyal bulguları ile her iki cinste de saptanan genital organ hipoplazileri ortak bulgulardır.

Schinzel-Giedion sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Güçlü bir zeka geriliği ile birlikte garip bir yüz yapısı ve çok sayıda doğumsal anomaliler görülür. Cüceliğe yakın düzeyde boy kısalığına neden olan genel gelişme geriliği vardır.

Bohring-Opitz sendromu, otosomal dominant yolla aktarılan, gelişme ve zeka geriliği bulgularının ön planda olduğu, hastaların çoğunun çocukluk yaşlarında kaybedildiği bir sendromdur. C sendromu'nun fenotipi olarak benimsenir.

Hartsfield sendromu, otosomal dominant yolla aktarılan, ender görülen, kalıtsal bir sendromdur. El ve ayak parmaklarının konjenital eksikliği (ektrodaktili), yarık dudak ve yarık damak ile kafatası malformasyonu (holoprosensefali) üçlüsü temel bulgulardır.
Osteopathia striata, kemik yoğunlaşmasının (osteoskleroz) saptandığı, X-kromozomu aracılığıyla (XLD) aktarılan kalıtsal bir sendromdur; olguların 1/3’ü sporadiktir.

van Buchem hastalığı, kemik yoğunlaşmalarıyla (osteoskleroz) karakterize, iki fenotipi olan kalıtsal bir hastalıktır. Fenotiplerden biri otosomal resesif yolla aktarılır ve kıyasla daha sık görülenidir. Otosomal dominant yolla aktarılan tipi ise çok ender görülür.

Hajdu-Cheney sendromu (arthrodentoosteodysplasia), iskelet sistemindeki defektlerle karakterize otosomal dominant yolla aktarılan kalıtsal bir sendromdur.
