22q11.2 deletion sendromu, kromozom anomalisi kökenlidir; olguların bir bölümünün kalıtsal olduğu saptanmıştır. Çok sayıda fenotipi vardır.

DiGeorge sendromu, 22q11 deletion içeren sendromlar grubunun bir fenotipidir. Olguların bir bölümü kalıtsaldır, otosomal dominant yolla aktarılır.Teratojenlerin ve diabetes mellitus'un neden olduğu mutasyonların da etkisi önemsenmektedir. Gebeliğin 4-7. haftasında beliren 3.-4. faringeal ark kompleksi etkilenmesine bağlı konjenital anomalilerle karakterizedir.
Opitz GBBB sendromu 22q11.2 sendromları kümesinin kalıtsal olan tek fenotipidir. 2 tip Opitz GBBB sendromu bilinmektedir.
Ağız-Yüz-Parmak sendromu , günümüze dek 16 fenotipi belirlenmiş olan bir sendromlar kümesidir. Bilinen fenotiplere yenileri eklenebilir. Ağız-Yüz-Parmak sendromunun OFD arasında en sık ratlanılanı OFP tip I temel bulguları içerir; sıkça rastlanan öteki fenotiplerde, tip I'e eklenen ya da çıkarılan yan bulgular vardır.
Ağız-Yüz-Parmak sendromu tip 6 , ektodermal displazi bulguları da içeren, otosomal resesif geçen, ender görülen kalıtsal bir sendromdur. Belirgin bir genel gelişme geriliği saptanır.

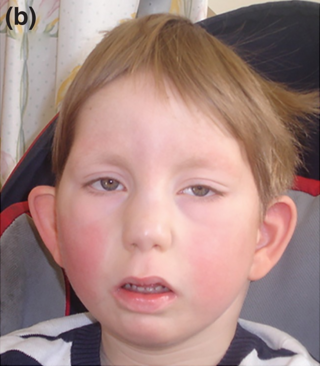
Baraitser-Winter sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; 2 fenotipi vardır. Genel gelişme geriliği izlenir. Kraniyosinostoz nedenli trigonosefali ve mikrosefali saptanır. Boyun kısadır. Hipertelorizm saptanır, kaş çıkıntıları yüksektedir. Göz kapakları iridir ve bilateral ptozis saptanır. Mikroftalmi ile iris ve retina defektleri (koloboma) izlenir. Kulak kepçeleri küçüktür ve aşağıdadır; işitme sorunları vardır. Burun sırtı kalın, ucu büyüktür. Uzun bir filtrum altında ince bir üst dudak bulunur. Alt dudak kalındır. Üst dudak ve damak yarıktır. Altçene geridedir (retrognati). Omurga sisteminde kifoz ve skolyoz türü malformasyonlar saptanır. Başparmak sayısı birden fazladır (polidaktili). Değişik oranlarda konjenital kalp anomalileri ve ürogenital anomaliler görülür. Beyinde corpus callosum anomalileri ile frontal lob anomalisi saptanır; epilepsi ve zeka geriliği bulguları vardır. Genel hipotoninin yanı sıra omuz kaslarında giderek artan güçsüzlük görülür. Deri lenfoması ve lösemi riski yüksektir.
Au-Kline sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur; böbrek, kalp ve yüz bulgularıyla öne çıkar.

Cornelia de Lange sendromu , 5 fenotipi olan kalıtsal bir sendromdur. Tüm fenotiplerde benzer bulgular saptanır; farklılık etkilenen genlerdedir.

Joubert sendromu, kalıtsal bir sendromdur; 36 fenotipinden 34'ü otosomal resesif yolla aktarılır, olguların bir bölümü Meckel sendromu ile çakışmaktadır. Joubert sendromunun tüm fenotiplerinde 3 temel bulgu vardır: santral sinir sistemi anomalileri, fizik gelişme geriliği, solunum sorunları. Serebellum (beyincik) vermisi ve beyin sapı hipoplazisi ile beyin radyolojisinde saptanan “azıdişi bulgusu ” santral sinir sistemi anomalilerinin başında gelir. Ataksi ve hipotoni sık görülür. Bazı hastalarda oksipital meningosel ya da meningomyelosel olabilir. Schinzel acrocallosal sendromu, önemli fenotiplerinden biridir.

Kabuki sendromu, iki fenotipi olan bir sendromdur: Kabuki sendromu 1 ve Kabuki sendromu 2 (Kabuk2). Kabuk1, otosomal dominant yolla aktarılan kalıtsal ya da izole olgular biçiminde görülür; Kabuk2 ise, X-kromozomu aracılığıyla dominant yolla (XLD) aktarılır. Her iki fenotipini çok sayıda ortak bulguları vardır; özellikle yüz, iskelet sistemi, gelişme geriliği, parmakizleriyle ilgili bulgular ve zeka geriliği önemlidir.
Pallister-Hall sendromu, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Hipotalamus'ta hamartoblastoma olarak nitelendirilen oluşumun yanı sıra çok sayıda endokrin sistem anomalileri, açılmamış anüs ve polidaktili bulguları ön plandadır. Hipofiz agenezi/hipoplazisi kökenli hipopituitarizm, adrenal gland hipoplazisi bulguları, hipotiroidizm, endokrin sistem anomalilerinin başlıcalarıdır.
Ritscher-Schinzel sendromu, 2 fenotipi olan kalıtsal bir sendromdur.

Toriello-Carey sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Doğum ölçümleri normaldir, ancak zamanla gelişme geriliği bulguları belirir.

Zunich nöroektodermal sendromu , otosomal resesif yolla aktarılan, glikozilfosfatidilinositol sentezi bozukluğuna bağlı kalıtsal bir sendromdur; gözlerde retinal kolobomalar, konjenital kalp defektleri, deri hastalıkları, kulak anomalileri ve zeka geriliği en sık rastrlanan bulgulardır.

Bohring-Opitz sendromu, otosomal dominant yolla aktarılan, gelişme ve zeka geriliği bulgularının ön planda olduğu, hastaların çoğunun çocukluk yaşlarında kaybedildiği bir sendromdur. C sendromu'nun fenotipi olarak benimsenir.

Hartsfield sendromu, otosomal dominant yolla aktarılan, ender görülen, kalıtsal bir sendromdur. El ve ayak parmaklarının konjenital eksikliği (ektrodaktili), yarık dudak ve yarık damak ile kafatası malformasyonu (holoprosensefali) üçlüsü temel bulgulardır.

Oculofaciocardiodental sendrom, göz malformasyonları, çene-yüz anomalileri, dolaşım sistemi ve bağışıklık sistemi defektleriyle karakterize, X-kromozomu aracılığıyla dominant (XLD) yolla aktarılan kalıtsal bir sendromdur. Mikroftalmi sendromunun 18 fenotipinden biridir.

Saethre-Chotzen sendromu (acrocephalosyndactylia III), fiziksel gelişme geriliği, kraniyosinostoz nedenli kafatası anomalileri, asimetrik yüz, göz ve parmak malformasyonlarının saptandığı otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Kraniyosinostozun çok sayıda eklemi etkilediği olgularda “kafaiçi basıncı artışı sendromu (KİBAS)” gelişebilir.

Shprintzen-Goldberg sendromu , marfanoid yapı, kraniyosinostoz, kardiyovasküler ve nörolojik anomalilerle karakterize, otosomal dominant yolla aktarılan kalıtsal bir sendromdur. Marfanoid yapı, Marfan sendromundaki bulguların büyük bölümünün saptandığı, ancak Marfan sendromunudaki genetik altyapının bulunmadığı olgular için kullanılan nitelemedir; uzun kollar ve bacaklar, örümceksi parmaklar (araknodaktili), eklemlerde aşırı gevşeklik ve mitral kapak prolapsusu Marfan sendromunu andıran başlıca bulgulardır. Robbins Shprintzen-Goldberg sendromu’nun, Marfan sendromu ve Loeys-Dietz sendromu ile oldukça çok büyük benzerlikleri vardır.
HMC sendromu, otosomal resesif yolla aktarılan kalıtsal bir sendromdur. Sendroma adını veren 3 temel bulgu vardır: Hipertelorizm-küçük kulak deliği (microtia)-yüz yarıkları. Mikrosefali ve hipertelorizm belirgindir. Kulak boşlukları dardır ve işitme güçlüğü vardır. Burun sırtı yayvandır. Ağız açıklığı küçüktür (mikrostomi); yüz, burun, dudak ve damak yarıkları bulunur. Konjenital kalp defektleri, böbreklerde yer değişikliği, vertebra anomalileri saptanır. Serçe parmağı kısadır, parmaklarda yapışıklıklar vardır. Psikomotor gelişme geriliği görülür.
