
Hamilelik veya gebelik, erkekten gelen sperm ile kadının yumurtalıklarından atılmış olan yumurtanın döllenmesi ile meydana gelen fetusun kadın organ ve dokularında değişiklikler meydana getirdiği, doğuma kadar geçen yaklaşık 9 aylık dönem.

Kromozom 18, toplamda 22 çift olan otozomal insan kromozomlarından onsekizincisidir. İnsanlarda normalde bir çift halinde bulunur. 76 milyon baz çiftine ve toplam hücre DNA'sının %2,5'ine sahiptir. Kromozom 18, muhtemelen 300 ile 400 arasında gen içermektedir.

Down sendromu, trizomi 21 ya da mongolizm; genetik düzensizlik sonucu insanın 21. kromozom çiftinde fazladan bir kromozom bulunması durumu ve bunun sonucu olarak ortaya çıkan genetik bir bozukluktur. Down sendromu, bireyin 1 yaşından daha uzun süre yaşayabildiği tek otozomal trizomidir.
İkili tarama testi ya da 11-14 testi olarak da bilinen ilk trimester tarama testi Down sendromu ve Trizomi 18 adı verilen kromozomal anomaliye sahip bebekleri gebeliğin çok erken dönemlerinde saptamaya yönelik bir tarama testidir. Yaşları kaç olursa olsun tüm kadınlar fiziksel veya zeka engelli bebek doğurma riski taşırlar. Down sendromuna sahip bir bebek doğurma riski 20 yaşındaki bir kadında 1530'da 1 iken bu risk artarak 44 yaşındaki bir kadında 30'da 1'e çıkar.
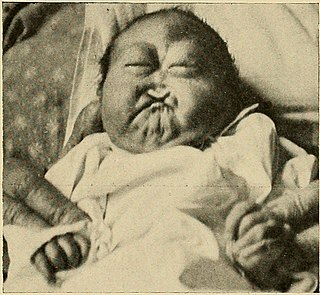
Patau sendromu ya da Trizomi 13, görülme sıklığı 10.000 canlı doğumda bir görülen bir kromozomal anomalidir.
İnsandaki nöral tüp adı verilen yapı beyin dokusundan başlayıp, boylu boyunca omuriliği de içine alacak şekilde aşağı doğru uzanan bir yapıdır. Bu yapı, döllenmeden sonraki 2. ile 3. hafta arasında gelişimini tamamlar. Gelişimin herhangi bir nedenle sorunlu olması, tüpün herhangi bir bölgesinin açık kalmasına neden olur ve bu duruma nöral tüp defekti (kusuru) adı verilir.
Üçlü tarama testi, down sendromu, nöral tüp defekti ve trizomi 18 adı verilen genetik hastalığın anne karnındaki bebekte olma olasılığını saptayan bir kan incelemesidir. Ayrıca üçlü test yardımı ile diğer bazı anomalileri de saptamak olasıdır. Bebeğin karın duvarı anomalilerinde, böbrek anomalilerinde de test sonuçları yüksek çıkabilir.

Smith-Lemli-Opitz (SLO) sendromu, otozomal resesif geçişli, çok sayıda doğuştan anomalinin gözlendiği nadir görülen bir sendromdur. Smith-Lemli-Opitz sendromlu olgularda kolesterol biyosentezinin son basamağında 7-dehidrokolesterolü (7DHK) kolesterole çeviren ve geni 11q13'te lokalize bir enzim olan 7DHK redüktazın doğuştan eksikliği söz konusudur. Görülme sıklığı 1/20.000 ile 1/60.000 arasında değişmektedir.
Artrogripozis, genellikle çoğul kongenital kontraktürleri tarif etmek için kullanılmaktadir. Kelimenin kökeni Yunancadan gelmektedir ve "bükük veya çengelleşmiş eklemler" anlamındadır. Bu kompleks sendrom çoğul eklem kontraktürleri, az gelişmiş ve kontrakte kaslar, ekstremite deformiteleri ve bunun yanında duyunun sağlam olması ile karakterizedir.

Teratoloji doğuştan gelen bozukluklar ya da kusurlar için kullanılan bir terimdir. Yunanca canavar ya da harika anlamına gelen τέρᾰς ile söz anlamına gelen λόγος sözcüklerinden gelmektedir. Aynı anlamda kullanılan bir başka terim de dismorfolojidir.
Konjenital bozukluk olarak da bilinen doğum kusuru, nedeni ne olursa olsun doğumda mevcut olan anormal bir durumdur. Doğum kusurları fiziksel, zihinsel veya gelişimsel engelliliklerle sonuçlanabilir.
Mikrognati (micrognathism), altçenenin yetersiz gelişmesi (hipoplazi) niteliğinde bir anomalidir; 1. ve 2. faringeal arklara (brankial yarık) özgü malformasyonların çoğu çene-yüz bölgesindeki mezenkimal dokunun embriyolojik dönemdeki yetersizliğine bağlanmaktadır. Altçene küçüktür ve geridedir; bu olgu, çevredeki kasların hareketlerini sınırlarken bu kaslardan yararlanan dokuların gelişmeleri de kısıtlanır. Kraniyofasiyal anomalileri içeren sendromlarda çok sık görülen bulgulardan biridir. Sendroma-özgü olmayan olguların varlığı da bilinmektedir; örneğin; altçenesi, kafa ile göğüs kafesi arasında sıkışmış olan bir fetüste çene kemiklerinin gelişimi olması gereken düzeye ulaşamamaktadır. Gebelik sorunları, gebelerin alkol kullanması, gestasyonel diabet (gebelik diabeti) gibi çevresel faktörler etkili olabilmektedir (Möbius sendromu). Olguların bir bömümü, gebelerdeki rubella (kızamıkçık) infeksiyonu komplikasyonudur. Yenidoğanların bir bölümünde görülebilen hafif mikrognatiler, altçene gelişiminin tamalanmasıyla silinebilir. Mikrognati'de altçene gövdesi kadar çene eklemini oluşturan yapıların hipoplazisi de önemlidir. Mikrognatilerin bir bölümü kulak sistemi, üstçene ve damak gelişiminin de aksadığı olgularla birlikte görülür.
TORCH, annedeki bir infeksiyon etkeninin, plasenta aracılığıyla ya da doğum sırasında bebeğe geçmesi olgusudur. TORCH tablosu ölü doğum, erken doğum ve prematüre bebek ölümlerinin önemli nedenlerinden biridir. İnfeksiyon etkenlerinin baş harfleriyle oluşturulan TORCH kısaltmasında yer alan canlı etkenlere giderek yenileri eklenmektedir.
Makrosomi, [macro+soma (σῶμα)] aşırı büyüme sonucu oluşan iri vücut, iri-yarı vücut yapısı, en ve boy ölçümleri normalden fazla vücut tanımı yapan terimdir; ilk kez 19. yüzyıl ortalarında, Dr. Robley Dunglison (1798–1869) tarafından yazılan kitapta kullanılmıştır. Aşırı büyüme, doğum öncesi (prenatal) dönemde ya da doğumdan sonra (postnatal) dönemde ortaya çıkabilen bir olgudur. Makrosomi olgularının bir bölümü “aşırı büyüme sendromları”nın özgün bir bileşenidir.
Multipl konjenital anomaliler-hipotoni-epilepsi sendromu, 3 fenotipi olan kalıtsal bir sendromdur. Fenotip 1 (MCAHS1) ve fenotip 3 (MCAHS3) otosomal resesif, tip 2 (MCAHS2) ise x-kromozomu aracılığı ile resesif olarak aktarılır. Birçok enzimin çalışabilmesinde, kompleman sisteminin düzenlenmesinde ve hücrelerarası iletişimin sağlanmasında önemli katkıları olan, fosfogliserid yapısındaki glikosilfosfatidilinositol sentezindeki sorunlarından kökenlidir. Nörolojik belirtiler sendromun fenotiplerindeki temel bulguları oluştururlar; bunlara eklenen çok sayıdaki anomalilerin yeri, sayısı ve gücü farklıdır.

Serebrokostomandibular sendrom , otosomal dominant yolla aktarılan kalıtsal bir sendromdur; çok az sayıda otosomal resesif olgular da vardır. Bebeklerin yarıya yakın bölümü 1 yaşından önce kaybedilir. Doğum ölçümleri normaldir, ancak zamanla gelişme geriliği bulguları belirir.

Zunich nöroektodermal sendromu , otosomal resesif yolla aktarılan, glikozilfosfatidilinositol sentezi bozukluğuna bağlı kalıtsal bir sendromdur; gözlerde retinal kolobomalar, konjenital kalp defektleri, deri hastalıkları, kulak anomalileri ve zeka geriliği en sık rastrlanan bulgulardır.

Klippel-Feil anomalisi, boyun omurlarını ilgilendiren konjenital bir anomalidir; kalıtsal olgular otosomal dominant yolla aktarılır. Klippel-Feil sendromu'nun temel anomalileridir. Başlıca 3 bulgu saptanır:
- Servikal vertebralarda hipoplazi ve kaynaşma
- Servikal vertebralardaki anomaliye bağlı olarak boyun kısa ve hareketleri sınırlıdır
- Trapez kaslarında(*) yelkensi görünüm saptanır.

Konjenital kalp defektleri ya da konjenital kalp hastalıkları, kalp işlevlerini olumsuz yönde etkileyen, sık görülen doğumsal patolojilerdir. Gebeliğin 3.-8. haftalarındaki embriyogenez kusurunun sonucudur. Doğumların %1’ında görülürler; prematürelerde ve ölü doğum bebeklerinde daha sıktır.

Konjenital amputasyon, bir uzuv veya uzuv olmadan veya uzuvların bir parçası olmadan doğumdur.