Dudak-damak yarığı
| Dudak damak yarığı | |
|---|---|
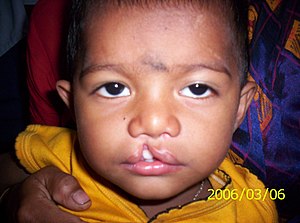 | |
| Dudak-damak yarıklı bir hasta | |
| Uzmanlık | Medikal genetik |
Dudak-damak yarığı (orofasiyal yarık), embriyolojik dönemde çeşitli nedenlerden dolayı bebeğin yüz bölgesindeki yapıların birleşme kusuru nedeniyle ortaya çıkan bir hastalıktır. Türkçede, halk arasında tavşan dudaklılık olarak bilinir.
Uterus içi yaşamda, fetüsün dudak yapısını oluşturan hücrelerin birleşmesi 4 veya 5. haftada, damak yapısını oluşturan hücrelerin birleşmesi ise 8 veya 9. haftada başlamaktadır. 12. haftanın sonunda, fetüsün damak ve dudak dokularının birleşmesi tamamlanmış olur. Birleşmenin tam olarak sağlanamaması durumunda fetüste oral yarıklar meydana gelir.[1][2]
Tipleri
Bazı hastalarda sadece yarık dudak veya yarık damak olmakla birlikte, bazılarında ise hem dudak hem de damak yarığı bir arada bulunabilmektedir.
- Oral yarıklar aşağıdaki tiplerde görülebilmektedir
- Primer (Ön) damak yarıkları
- Sekonder (Arka) damak yarıkları
- Hem primer hem de sekonder damak yarıkları
- Tek taraflı yarık dudak
- İki taraflı yarık dudak
- Medyan yarık dudak
Oral yarığın ağız boşluğuna uzanan kısmı bazı durumlarda mukoza ile kaplı olabilmekte ve buna sub-mukozal damak yarığı denmektedir.[2]
Oluşum nedenleri
Oral yarık sıklığı yaklaşık 1/700 olarak bildirilmiştir. Siyah ırkta oran daha düşük, Kızılderililer, Japonlar ve Çinlilerde ise daha fazladır. Yarık dudak vakasının sıklığı 1/300, sadece yarık damak 1/1500 ve her iki vakanın bir arada bulunma sıklığı da 1/2500 olarak bildirilmiştir. Finlandiya'da yarık damak sıklığı 1/1000, kuzey-doğu Fransa'da 0,41/1000, İtalya'da 0,34/1000 olarak görülmüştür. Oranların bu kadar farklı olmasının nedenleri arasında coğrafi, etnik köken ve kalıtım kalıplarının farklılıkları olabileceği gibi oral yarığın tipine göre de farklılık göstermektedir. Yarık dudaklı hastaların %50'sinde yarık damak bulunmaktadır.[1][3][4]
Oral yarıkların oluşumuyla ilgili yapılan birçok çalışma olmasına rağmen etyolojisi ve patojenitesi hala tam anlamıyla açıklığa kavuşmasa da yarık damak/ dudak oluşumunun genetik ve çevresel faktörlerin etkileşimiyle meydana geldiği bilinmektedir. Kimi vakalarda sadece genetik veya çevresel faktörler olabildiği gibi bazı vakalarda hem genetik hem de çevresel faktörler birlikte etkili olmaktadır. Tek yumurta ikizlerinde yarık damak/ dudak oluşumunun %100 olmaması genetik faktörlerin tek başına etkili omadığını göstermektedir.[4] Gebelik döneminde anti-epileptik ilaç, içki ve sigara kullanımı; folik asit ve multivitamin eksiklikleri gibi genetik olmayan çevresel faktörlerin yarık damak/ dudak ile ilişkisi ortaya konmasına rağmen hastalığın genetik temeline dayanan çalışmalar ön plana çıkmıştır.[3][5] İzole damak yarıklı dominant kalıtım tipi gösteren aileler olmasına rağmen yarık damaklı vakaların çoğu Mendelyen kalıtım tipi (tek gen hastalıkları) göstermez. Curtis ve arkadaşları 1961 yılında yaptıkları çalışmada bir çocukta yarık damak varsa diğer çocukta oluşma sıklığı %2, ebeveynlerden birinde varsa %6, hem ebeveynlerden biri hem de bir çocukta varsa oran %15 olarak bildirmiştir.[6] Tek yumurta ikizlerinde oran %40 ile %60, çift yumurta ikizlerinde %5 olarak bildirilmiştir.[4] Bugüne kadar yapılan çalışmalarda, İngiliz toplumunda,[7] Çin kökenli ailelerde,[8] Kuzey ve Güney Amerikalı ailelerde[9] ve iki geniş Suriyeli ailede[10] geniş genom taramaları yapılmış, aday genler ve kromozom haritaları çıkarılmıştır.[11][12][13]
Palatogenezis (damak oluşumu) birçok sinyal molekülü ve transkripsiyon faktörlerini kapsayan, fetal hayatın erken evrelerinde gerçekleşen metabolik bir olaydır. Bu metabolik yolaktaki moleküllerden birinin eksikliği veya aktivite sorunu oluşmakta olan oral dokularda kusurlara neden olmaktadır. Palatogenezis sürecinde metabolik yolağı etkileyebilecek herhangi bir mutasyon veya kromozomal sorun sadece oral yarık oluşumuna neden olmayıp başka sendromlarında ortaya çıkmasına neden olabilmektedir. Sendromik vakalarda yapılan çalışmalar asendromik vakalara göre çok daha fazla olsa da oral dokuların kusuruna neden olan faktörlerin aydınlatılmasında yeterli olamamıştır.
Bugüne kadar yapılan genetik çalışmalar, sadece aday genlerin salt analizleriyle değil aday genlerin birbirleriyle bağlantılarına da yönelik olmuştur. Aday genler arasında en çok çalışılanı TGFα (Transforme edici Büyüme Faktörü alfa), MSX1, TGFβ (Transforme edici Büyüme Faktörü beta), BCL3, IRF6 gibi genlerdir.[1][3][3][4] Kimi araştırmacılar bu genler ile hastalık arasında positif, kimileri de negatif ilişki bulmuştur. Positif veya negatif ilişkiyi sedece genler direkt olarak değil, çalışmanın yapıldığı popülasyon ve dış faktörler de etkilemektedir. Bu genler farklı kalıtım kalıpları gösterdiklerinden (otozomal dominant, otozomal resesif gibi) oral yarık insidans aralığı geniştir. Bir genin diğeri üzerine etkisi düşünüldüğünde çok genli kalıtım modeli gösteren ve çevresel şartlardan etkilenen oral yarık oluşumunun ne denli komplike bir sorun olduğu ve genetik temelinin anlaşılmasının ise çok daha uzun zaman alacağı tahmin edilmektedir.[1][11] Günümüze kadar yapılan genetik çalışmaların çoğunu Avrupalı ve beyaz Amerikalı popülasyonlar oluşturmaktaydı. Son 2-3 yılda, Çin, Kuveyt, Suriye, Hindistan gibi ülkeleri de kapsayan geniş popülasyon çalışmaları yapılmıştır.[10][12][14] Bu bölgelerdeki oral yarık insidansının yüksek çıkması genetik sonuçların daha da anlamlı olmasını sağlamıştır. Halen aday genler ile oral yarıkların arasında net bir bağlantı bulunamamıştır. Hastalığın meydana gelmesinde birden fazla gen, hatta gen gruplarının çevre faktörleri ile etkileşimi düşünülünürse daha fazla popülasyon taramalarının yapılmasına gerek vardır. Türkiye'de ise, az sayıda istatistiksel çalışma bulunmaktadır. Örneğin, Gazi Üniversitesi hastanesinde 1988-2005 yılları arasındaki 17.259 canlı doğumların 5/10.000'inde oral dudak-damak yarığı saptanmıştır. Hacettepe Tıp Fakültesinde yapılan başka araştırmada 1.229 yarık damak- dudak vakasının %19'unu izole dudak, %35,6'sını izole damak, %45'ini hem dudak hem damak yarığı oluşturmuştur.[15] Ancak, her iki çalışmada vakaların genetik temellerine dayalı bilgiler bulunmamaktadır. Genetik analiz temeline dayalı çalışmalar sadece vaka bazında yapılmış olsa da, büyük aile veya popülasyon çalışmalarına ihtiyaç vardır.
Galeri
Bu anomali, bir veya daha çok operasyon ile düzeltilebilmektedir.
 6 aylık bir kız bebeğin düzeltme operasyonunundan önceki durumu.
6 aylık bir kız bebeğin düzeltme operasyonunundan önceki durumu. Aynı kızın operasyondan bir ay sonraki durumu
Aynı kızın operasyondan bir ay sonraki durumu Aynı kızın 4,5 yaşındaki durumu. Operasyondan sonra yara izinin yaş ilerledikçe belirginliğini kaybettiği gözlemleniyor.
Aynı kızın 4,5 yaşındaki durumu. Operasyondan sonra yara izinin yaş ilerledikçe belirginliğini kaybettiği gözlemleniyor.
Kaynakça
- ^ a b c d Stainer P, Moore GE.: Genetics of cleft lip and palate: syndromic genes contribute to the incidance of non- syndromic clefts. Human Molecular Genetics 13, Review Issue 1, 2004
- ^ a b Moore K.L., Persaut T.V.N: İnsan Embriyolojisi, 6. baskıdan çeviri, Ed: Yıldırım M, Okar İ., Dalçık H. Nobel Kitabevleri, 2002.
- ^ a b c d Wong FK, Hagg U.: An update on the aetiology of orofacial clefts. Hong Kong Med J, 10: 331-6, 2004
- ^ a b c d Murray JC.: Gene/ environment causes of cleft lip and/ or palate.: Clin. Gen. 61:248- 256, 2002.
- ^ Wyszynski DF, Beaty TH.: Review of the role of potential teratogens in the origin of human nonsyndromic oral clefts. Teratology, May, 53(5): 305-17, 1996.
- ^ Curtis E.J., Fraser F, Warburton D.: Congenital cleft lip and palate. Am. J. Dis. Child.:102; 853- 57, 1961
- ^ Prescott NJ, Lees MM, Winter RM, Malcolm S.: Identification of susceptibility loci for nonsyndromic cleft lip with or without cleft palate in a two stage genome scan of affected sib-pairs. Hum. Genet., Mar,106(3):345-50, 2000.
- ^ Marazita ML, Field LL, Cooper ME, Tobias R, Maher BS, Peanchitlertkajon S, Liu YE.: Genome scan for loci involved in cleft lip with or without cleft palate, in Chinese multiplex families. Am. J. Hum. Gen., 71, 349- 364, 2002.
- ^ Zeiger JS, Hetmanski JB, Beaty TH, VanderKolk CA, Wyszynski DF, Bailey-Wilson JE, de Luna RO, Perandones C, Tolarova MM, Mosby T, Bennun R, Segovia M, Calda P, Pugh EW, Doheny K, McIntosh I.: Evidence for linkage of nonsyndromic cleft lip with or without cleft palate to a region on chromosome 2. Eur. J. Hum. Genet. Nov;11(11):835-9, 2003.
- ^ a b Wyszynski DF, Albacha-Hejazi H, Aldirani M, Hammod M, Shkair H, Karam A, Alashkar J, Holmes TN, Pugh EW, Doheny KF, McIntosh I, Beaty TH, Bailey-Wilson JE.: A genome-wide scan for loci predisposing to non-syndromic cleft lip with or without cleft palate in two large Syrian families. Am. J. Med. Genet. Dec 1;123(2):140-7, 2003.
- ^ a b Suazo J, Santos JL, Carreno H, Jara L, Blanco R.: Linkage disequilibrium between MSX1 and non-syndromic cleft lip/palate in the Chilean population. J Dent Res.,Oct;83(10):782-5, 2004.
- ^ a b Marazita ML, Field LL, Cooper ME, Tobias R, Maher BS, Peanchitlertkajorn S, Liu YE.: Nonsyndromic cleft lip with or without cleft palate in China: assessment of candidate regions. Cleft Palate Craniofac J., Mar;39(2):149-56, 2002.
- ^ Moreno LM, Arcos-Burgos M, Marazita ML, Krahn K, Maher BS, Cooper ME, Valencia-Ramirez CR, Lidral AC.: Genetic analysis of candidate loci in non-syndromic cleft lip families from Antioquia-Colombia and Ohio. Am J Med Genet A.,Mar 1;125(2):135-44, 2004.
- ^ Field LL, Ray AK, Cooper ME, Goldstein T, Shaw DF, Marazita ML.: Genome scan for loci involved in nonsyndromic cleft lip with or without cleft palate in families from West Bengal, India. Am. J. Med. Genet. A., Oct 15;130(3):265-71, 2004.
- ^ Tunçbilek G, Özgür F, Balcı S: Yarık dudak ve damak hastasında görülen ek malformasyon ve sedromlar. Çocuk sağlığı ve hastalıkları dergisi, 47: 172- 176, 2004.
Dış bağlantılar
- Konuyla iligli Fırat Üniversitesi Tıp Dergisinde çıkmış bir makale 4 Mart 2016 tarihinde Wayback Machine sitesinde arşivlendi., 28 Temmuz 2008 tarihinde erişildi.
| Sınıflandırma | |
|---|---|
| Dış kaynaklar |








