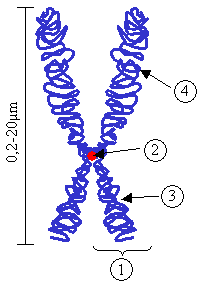
Down sendromu
| Down sendromu | |
|---|---|
| Sınıflandırma ve dış kaynaklar | |
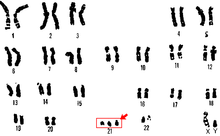 21 nolu trizomi karyotip | |
| ICD-10 kodu | Q90 |
| ICD-9 kodu | 758.0 |
| MedlinePlus | 000997 |
| eMedicine | ped/615 |
| OMIM | 190685 |
Down sendromu, trizomi 21 ya da mongolizm; genetik düzensizlik sonucu insanın 21. kromozom çiftinde fazladan bir kromozom bulunması durumu ve bunun sonucu olarak ortaya çıkan genetik bir bozukluktur.[1][2][3] Down sendromu, bireyin 1 yaşından daha uzun süre yaşayabildiği tek otozomal trizomidir.
Down sendromu vücutta yapısal ve fonksiyonel değişiklikler ile karakterize edilir. Vücuttaki küçük ve büyük farklılıkların kombinasyonu yapısal olarak sergilenir ve sık sık zihinsel kavramadaki bozukluklar ve fiziksel gelişimin tipik yüz görünümü gibi farklı olmasıyla ilişkilendirilir. Çoğunlukla hafif veya orta seviyeli öğrenme güçlüğü gibi sorunlar taşır.[4][5]
Down sendromu gebelik sırasında ya da doğumda tanımlanabilen bir rahatsızlıktır ve her 800 ile 1000 doğumda 1 oranında rastlanır. İstatistikler anne yaşının artışıyla bu oranın yükseldiğini göstermiştir, diğer etkenlerin payı küçüktür.
Down sendromunun tipik yüz siması, normal kromozom sayısına sahip olan bazı insanlarda da görülebilir. Ancak Down sendromunda buna ek olarak; el ayasında çift yerine tek derin olarak bulunan avuç içi çizgisi, epikantik katlanmanın neden olduğu badem biçimli göz, palebral yarık, düşük kas tonusu, ayak başparmağıyla ikinci parmak arası daha büyük bir boşluk ve sarkık dil morfolojisi görülebilir. Bu semptomların hepsi aynı anda görünmeyebilir, bazılarına sık rastlanırken bazıları daha seyrek görülür. Down sendromunun sağlığa getirdiği sorunların başında ise konjenital kalp defektleri ve kalp yetmezliği riskleri, gastroözafagal reflü hastalığı, tekrarlayan kulak enfeksiyonları, obstrüktif uyku apnesi ve tiroid bozuklukları riskleri sayılabilir.
Çocukluğun erken dönemlerinde sağlanacak olan aile ve tıp desteği ile erken müdahale sayesinde Down sendromlu insanlar destekle toplumla bütünleşik bir hayat kurabilirler.
Down sendromunun tanımlanışı
Down sendromunun adı, sendromu ilk kez 1866'da tanımlayan İngiliz hekim John Langdon Down'dan gelir. 1959'da Jérôme Lejeune tarafından 21. kromozomun trizomisi olduğu tanımlanmıştır.
Görünümleri

Bu bebekler doğduklarında farklı bir yüz görünümleri vardır.[3][5][6][7] Başları ufak, artkafa yassı, ense kısa ve geniştir. Burun kökü yassı, kulaklar kafada normalden düşük bir seviyede durur ve gözler birbirinden ayrık ve çekik görünür. (Bu görünüm Moğol ırkına benzetilerek mongolizm olarak da ifade edilir.) Dil, normal konuşmayı önleyecek kadar genişlemiştir. Ensede genellikle boğumlar vardır. Bu bebeklerin tonusları (vücut gerginliği) düşüktür. Geniş el, kısa ve tombul parmak ve sıklıkla avuç içlerinden birinde ya da ikisinde "Simian çizgisi" denilen tek bir çizgi vardır. Ellerin serçe parmakları genellikle içe doğru kıvrımlıdır. Vücut kısa ve tıknazdır. Çocukluk dönemlerinde solunum hastalıkları, kalp bozukluklarına rastlanabilir. Yaşam süreleri geçmiş yüzyılda düşük seyretmişken günümüzde gelişen tıp ve iyi bakım sonucunda bu yaş ortalama Down sendromlu kişi ömrü 50 yaş civarında seyretmektedir.
Down sendromunun getirilerinden biri de hafif ila orta düzey arasında değişebilen zeka geriliğidir.[4][8] Bu oran Mozaik Down sendromunda (açıklaması aşağıda) 10-30 oranında yukarıdadır.[8]
- Beyin sapı (özellikle pons) hipoplazisi
- Fontaneller açık
- Hipotoni (Moro refleksi güçsüz)
- Eklemlerde gevşeklik
- Boyun kalın-kısa
- Ense derisi kalın
- Basık yüz
- Orbitalar düz
- Gözler çekik
- Epikantus
- Kulak kepçesi anomalileri
- Pelvis displazisi
- El parmaklarında duruş anomalileri
- Konjenital kalp defektleri
- Mikrosefali
- Brakisefali
- Hipertelorizm
- Çekik göz kapakları
- Epikantus
- Strabismus
- Katarakt
- Displastik-lobülsüz kulaklar
- Basık yüz
- Yüz orta bölüm hipoplazisi
- Kafatası-yüz sinüsleri hipoplazisi/aplazisi
- Ağız açık
- Dilde oluklar (skrotal dil)
- Yarık dudak
- Yarık damak
- Sert damak kısa
- Dar çene yapısı
- Maloklüzyon
- Alveol kretleri kalın (üstçene lateral)
- Dişlerin gelişmesinde gerilik
- Dişlerin sürmeinde gecikmeler
- Sürekli dişlerde hipodonti
- Hipodonti+süpernümerer diş (hipohiperdonti)
- Mine hipoplazisi
- Taurodontism
- Periodontal patolojiler
- Parotis yetersizliği (hipoplazi)
- İskelet yaşı geriliği
- Toraks anomalileri
- Umblikal herni
- Reflü
- Gluten duyarlılığı (celiac)
- Duodenum ve anüs atrezisi
- Küçük penis
- İnmemiş testis
- Kısa ve yayvan eller
- Brakidaktili
- Serçe parmak kısa
- Avuçiçinde tek enine çizgi (simian line)
- Eklemlerde gevşeklik
- Servikal vertebra eklemleşme sorunları
- Deride hiperkeratoz ve seboreik keratoz bulguları
- Lösemi riski
- Entelektüel yetersizlik
- Alzheimer hastalığı riski
- Erişkinlerde serebrovasküler ataklar/felç
- İmmun sistem aksamaları
- Enfeksiyonlara duyarlılık
Gelişimleri
Down sendromlu çocuklara ek olarak zihin yetersizliği eşlik eder. genelde boy ve kilo açısından daha yavaş büyürler, daha yavaş öğrenirler, problem çözmede ve karar vermede diğer çocuklardan daha çok zorlanırlar. Zeka seviyeleri normalden düşük olarak kalır. Ancak iyi ve erken başlanan eğitimle zeka seviyelerinde anlamlı yükselmeye rastlanır. Down Sendromlu çocuklar iyi bir eğitimle normal birey şeklinde hayatlarını sürdürebilirler. İmkân tanındığında meslek edinebilirler. Kendi yaşamlarını idame ettirebilecek seviyeye ulaşabilirler. Fizik tedavi, özel eğitim ve dil terapisine ihtiyaç duyulur.
Özel eğitim

Down sendromlu çocuklar kendi aralarında farklılıklar gösterebilirler, bu yüzden çocuğun ihtiyaçlarına uygun bir programla özel eğitim, beraberinde sosyal ve duygusal gelişimi, bilişsel gelişimi ve motor gelişimi desteklenir.[4]
Fizik tedavi
Fizik tedaviye Down sendromlu bebeklerde iki aylıkken başlanmalıdır.Egzersizler fizyoterapist bakımında yapılmalı ve günlük programlarla evde aile tarafından uygulanmalıdır. Düzenli kontrollerle duruma göre tedavi desteklenir. Çocuklarda yüz kasları gevşektir. Fizik tedavi süresince kas gücü ve motor becerilerinin yanı sıra, algılama becerisi de programa dahil edilerek desteklenmelidir.
Dil terapisi
Down sendromlu çocuklarda konuşma geç gelişir. Erken dönemde başlanan dil terapisi ile ortalama 2-3 yaşında konuşma başlayabilir. Nadir rastlansa da bazıları çok geç konuşurlar.Hiç konuşamayan sayısı ise oldukça azdır.
Down sendromunun nedenleri
Sağlıklı bir insanın vücudundaki her hücrede 46 tane kromozom vardır. Oysa Down sendromlu bebeklerin hücrelerinde toplam 47 kromozom bulunur. Karyotipleri 47, XX+21 (dişi) ya da 47, XY+ 21 (erkek) şeklinde gösterilir. Yani fazladan bulunan kromozom vücut kromozomlarının yanında bulunur. Bu kromozom fazlalığının neden kaynaklandığı tam olarak bilinmese de, 35 yaşından sonra doğum yapan kadınların çocuklarında görülme olasılığı yüksektir. Bunun nedeni kromozom ayrılmalarının ileri yaşlarda daha düzensiz olmasından kaynaklanmaktadır. Bununla beraber, hücre bölünmesi sırasında meydana gelen ayrılmamalar da bu hastalığın sebeplerinden olabilir.
Down sendromunun epidemiyolojisi her canlı 800-1000 doğumda 1 Down sendromlu doğum oran olduğunu göstermiştir.
Down sendromu tipleri
Trizomi 21
Trizomi 21 (47, XX,+21); mayoz bölünme sırasında meydana gelen ayrılmama durumuyla ortaya çıkan fazla 21. kromozomun sebep olduğu Down sendromu tipidir. Yumurta ya da spermde bulunan fazla 21 ile bir gametde toplam 24 kromozom bulunur. Down sendromunun yaklaşık %95'ini kapsayan en çok görülen tipidir.
Mozaisim
Trizomi 21'in vücut hücrelerinin bazılarında görülmesi, bazılarında ise görülmemesi durumudur. Karyotip (46, XX/47, XX,+21) şeklinde gösterilip, hastalık "Mozaik Down Sendromu" olarak adlandırılır. Hastalık, mozaismin yoğunluğuna göre farklı seyredebilir. Trizomi 21 oranı ne kadar çok ise, çocuk Down sendromu özelliklerini o kadar çok gösterir. Mozaik Down sendromu, %1-2 oranında bir yere sahiptir.
Robertsonian tip translokasyon
Down sendromunda fazla 21. kromozom bazen Robetrsonian tip translokasyon ile görülür. Burada genellikle 21. kromozomun uzun kolu başka bir kromzoma bağlanır. Bu durumda karyotip 46, XX, t(14;21) şeklinde gösterilmekte fakat 14. kromozomda transloke olmuş bir 21. kromozom bulunmaktadır. Ya da izokromozom olarak da iki 21. kromozomun translokasyonu ile de Down sendromu 45, XX, t(21q;21q) şeklinde meydana gelebilir. Robertsonian tip translokasyon ile olan Down sendromları, toplam Down sendromunda %2-3'lük bir paya sahiptir.
21. kromozomun çift olması
Nadir olarak, 21. kromozomun duplikasyonu (kendini eşlemesi) ile de Down sendromu görülebilir. Burada 21. kromozom tam olarak bütün genleri taşımasa da, parça şeklinde görülür ve hastalığı tanımlar. Karyotip, (46, XX, dup(21q)) şeklindedir.
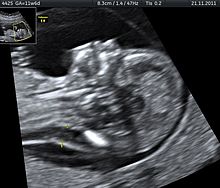
Doğum öncesi tanı
Down sendromu gebelikte tanınabilen bir genetik farklılıktır. İkili tarama testi, üçlü tarama testi, ultrasonografi ve diğer bazı tanı yöntemleri ile Down sendromundan şüphelenilen gebeliklerde ileri tetkikler yapılır. Özellikle ikili test sayesinde, doğru ölçülen bir ense kalınlığı ve burun kemiği ile birlikte down sendromlu bebeklerin %93'ünü tespit etmek mümkündür. Bu oran üçlü, dörtlü ve ardaşık testlerde % 75-80 dolaylarındadır.[12][]
CVS veya amniyosentez ile kesin tanı konur. Down sendromu saptanmışsa aileye ayrıntılı genetik danışmanlık verilir. Günümüzde geçerli olan uygulamada anne-babanın aile bütünlüğüne ve kişisel kararlara saygı çerçevesinde, bu tanıyı ileten hekimin gebeliğin devamı veya sonlandırılması konusunda yorumda bulunmaması gereklidir.
Ayrıca bakınız
Kaynakça
- ^ "Trisomy 21: The Story of Down Syndrome paper". 19 Mayıs 2015 tarihinde kaynağından arşivlendi.
- ^ a b c Lubec G, Engidawork E. The brain in Down syndrome (TRISOMY 21). Journal of Neurology, 249(10):1347-1356, 2002
- ^ a b c Roizen NJ, Patterson D. Down's syndrome. Lancet, 361(9365):1281-1289, 2003
- ^ a b c d e Capone G, Goyal P, Ares W, Lannigan E. Neurobehavioral disorders in children, adolescents, and young adults with Down syndrome. American Journal of Medical Genetics Part C: Seminars in Medical Genetics, 142C(3):158-172, 2006
- ^ a b c Hall, B. (1964). Mongolism in newborns: a clinical and cytogenetic study. 154. Acta Paediatrica Scandinavica. ss. 1-95.
- ^ a b DeLuke DM, Haug RH. Syndromes of the Head and Neck. Elsevier, Philadelphia, 2014
- ^ a b Steingass KJ, Chicoine B, McGuire D, Roizen NJ. Developmental disabilities grown up: Down syndrome. Journal of Developmental & Behavioral Pediatrics, 32(7): 548-558, 2011
- ^ a b "Keep Kids Healthy article on Down syndrome". 10 Eylül 2013 tarihinde kaynağından arşivlendi. Erişim tarihi: 3 Haziran 2006.
- ^ Haritha A, Jayakumar A. Syndromes as they relate to periodontal disease. Periodontology 2000, 56:65–86, 2011
- ^ Gimenez-Barcons M, Casteras A, Armengol MP, et al. Autoimmune predisposition in Down syndrome may result from a partial central tolerance failure due to insufficient intrathymic expression of AIRE and peripheral antigens. Journal of Immunology, 193: 3872-3879, 2014
- ^ Chadi MJ, Saint Georges G, Albert F, et al. Major salivary gland aplasia and hypoplasia in Down syndrome: review of the literature and report of a case. Clinical Case Reports, 5(6):939-944, 2017
- ^ "Arşivlenmiş kopya". 1 Ekim 2016 tarihinde kaynağından arşivlendi. Erişim tarihi: 28 Eylül 2016.